

 PDF
PDF Citation
Citation Print
Print



Introduction
Colorectal cancer (CRC) ranks as the third most common cancer globally and in the Republic of Korea [1,2]. Advancements in next-generation sequencing (NGS) have provided profound insights into CRC’s genomic alternations [3]. Typically, CRC is categorized as hypermutated or non-hypermutated, with the latter constituting over 90% of cases and frequently featuring alternations in the APC, TP53, and KRAS genes. In metastatic CRC (mCRC), standard treatment regimens now include EGFR, VEGF, BRAF, and HER2 targeting agents [4-6], with promising results from clinical trials involving KRAS-targeting drugs [7]. Yet, the median survival for mCRC patients remains at approximately 24 to 30 months.
The limited prognosis in mCRC stems from acquired resistance to anti-cancer agents due to the clonal evolution of cancer cells [8]. Somatic mutations specific to cancer can be categorized as clonal or subclonal, where subclonal diversity progressively increases during cancer progression and metastasis [9]. Therefore, a comprehensive and longitudinal genomic characterization of tumor DNA at baseline and progressive disease (PD) is increasingly vital in deciphering resistance mechanisms.
While conventional clinical genomic testing primarily uses tumor tissue DNA (ttDNA), circulating tumor DNA (ctDNA) profiling emerges as an attractive alternative in oncology [10]. Extensive literature covers the potential clinical utility of ctDNA [11]. Unique molecular identifiers (UMIs) have notably reduced the minimum variant allele frequency (VAF) detection limit to 0.1% by mitigating sequencing background error rates. This is achieved by assigning a UMI to each DNA template, followed by uniquely tagged template amplification to create UMI families [12]. A recent study by Kim et al. [13] compared ctDNA at baseline (baseline-ctDNA) and ctDNA at PD (PD-ctDNA) in patients with mCRC undergoing various lines of palliative systemic anti-cancer therapy. However, since this study did not compare the conventional cancer panel results using ttDNA samples, whether the genomic landscape identified by ctDNA sequencing precisely mirrors that of ttDNA sequencing remains uncertain.
In this study, we aimed to assess the clinical utility of ctDNA compared to ttDNA profiling. For each patient, we analyzed four types of DNA: ttDNA, germline DNA (gDNA), baseline-ctDNA, and PD-ctDNA. Our goal was to determine if UMI-based ctDNA profiling detects genomic alterations at frequencies comparable to those obtained by ttDNA sequencing. Additionally, we aimed to explore the clinical applicability of ctDNA and to better understand clonal evolution during anti-cancer therapy.
Materials and Methods
1. CRC patient cohort
All patients were recruited from Seoul National University Bundang Hospital (SNUBH), a tertiary hospital in the Republic of Korea. Each patient provided DNA from four different sources: ttDNA from tumor tissue, gDNA from blood, baseline-ctDNA, and PD-ctDNA. We conducted targeted sequencing for all liquid specimens. Tissue specimens were obtained from primary or metastatic tumors via resection, endoscopic, or needle biopsy. For patients with available tumor NGS data in the electronic medical records (EMR), we extracted genomic testing results, including a curated list of crucial mutations with details such as functional annotations (e.g., amino acid changes) and VAFs. Patients without accessible tumor NGS data underwent additional targeted sequencing using tumor tissues (described below).
2. Statistical analysis of survival data
Treatment response was evaluated based on the Response Evaluation Criteria in Solid Tumor (RECIST) ver. 1.1 [14]. Progression-free survival (PFS) was calculated from the start date of treatment to the date of documented disease progression, relapse, or any cause of death, whichever occurred first. Overall survival (OS) was determined from the start date of treatment to the date of death from any cause. Survival outcomes were computed using the Kaplan-Meier method and compared with the log-rank test. Optimal VAF cutoff values for OS were identified using maximally selected rank statistics [15] in the ‘maxstat’ R package. Two-sided p-values below 0.05 were considered statistically significant. Clinical data were analyzed using IBM SPSS Statistics ver. 26 (IBM Corp., Armonk, NY) or R ver. 4.3.1 (R Foundation for Statistical Computing, Vienna, Austria).
3. Sample preparation
The plasma was isolated within 30 minutes of blood collection to prevent the ctDNA degradation and gDNA release from white blood cells. Each sample underwent centrifugation with Ficoll-Paque Plus (Cytiva, Marlborough, MA) at 2,000 rpm for 15 minutes without interruption. Before further processing, careful examination ensured plasma quality. Subsequently, an extra centrifugation stage was carried out at 16,000 ×g for 15 minutes at 4°C to eliminate cell debris. Following this, 1 mL aliquots of plasma in 1.5 mL tubes were stored at –80°C until extraction. The ctDNA extraction from plasma was carried out using the QIAamp Circulating Nucleic Acid Kit (Qiagen, Valencia, CA). Additionally, gDNA was isolated from peripheral blood mononuclear cells using the QIAamp DNA Mini Kit (Qiagen).
4. Next-generation sequencing
DNA from liquid specimens (i.e., gDNA, baseline-ctDNA, and PD-ctDNA) was sequenced with Axen Cancer Panel 1 (ACP1) (Macrogen, Inc., Seoul, Korea), which consists of 88 cancer-associated genes (S1 Table). For ctDNA, we utilized UMI tags to reduce the rate of false-positive variant calls and increase the sensitivity of variant detection. In patients without tumor NGS data in EMR, their tumor tissues were also sequenced with Axen Cancer Master Panel (ACMP) (Macrogen, Inc.), which consists of 558 cancer-associated genes (S1 Table). Both ACP1 and ACMP libraries were prepared for sequencing using the SureSelectXT Library Prep Kit (Agilent Technologies, Inc., Santa Clara, CA) per the manufacturer’s recommendations. Sequencing was performed with the NextSeq 500 system (Illumina, Inc., San Diego, CA) using 150 base pairs (bp) paired-end reads. Raw sequencing data were produced in the Binary Base Call (BCL) file format, which was then converted to individual FASTQ files by demultiplexing with the bcl2fastq software (v2.19.0) (Illumina). Additionally, FASTQ files from ctDNA were split to trimmed sequence reads and molecular barcodes using the Trimmer program from the Agilent Genomics NextGen Toolkit (AGeNT) ver. 3.0 (Agilent Technologies). Next, sequence reads in FASTQ files were mapped to the Human Genome version 19 (hg19) reference genome using the BWA-MEM software, producing binary alignment map (BAM) files. The BAM files from ctDNA underwent error correction through UMI deduplication using the LocatIt program from AGeNT. Finally, all BAM files were recalibrated for potential systematic biases that could affect the assignment of base quality scores by the sequencer using the ApplyBQSR command from the Genome Analysis Toolkit (GATK) ver. 4.2.5.0 [16].
5. Somatic variant discovery
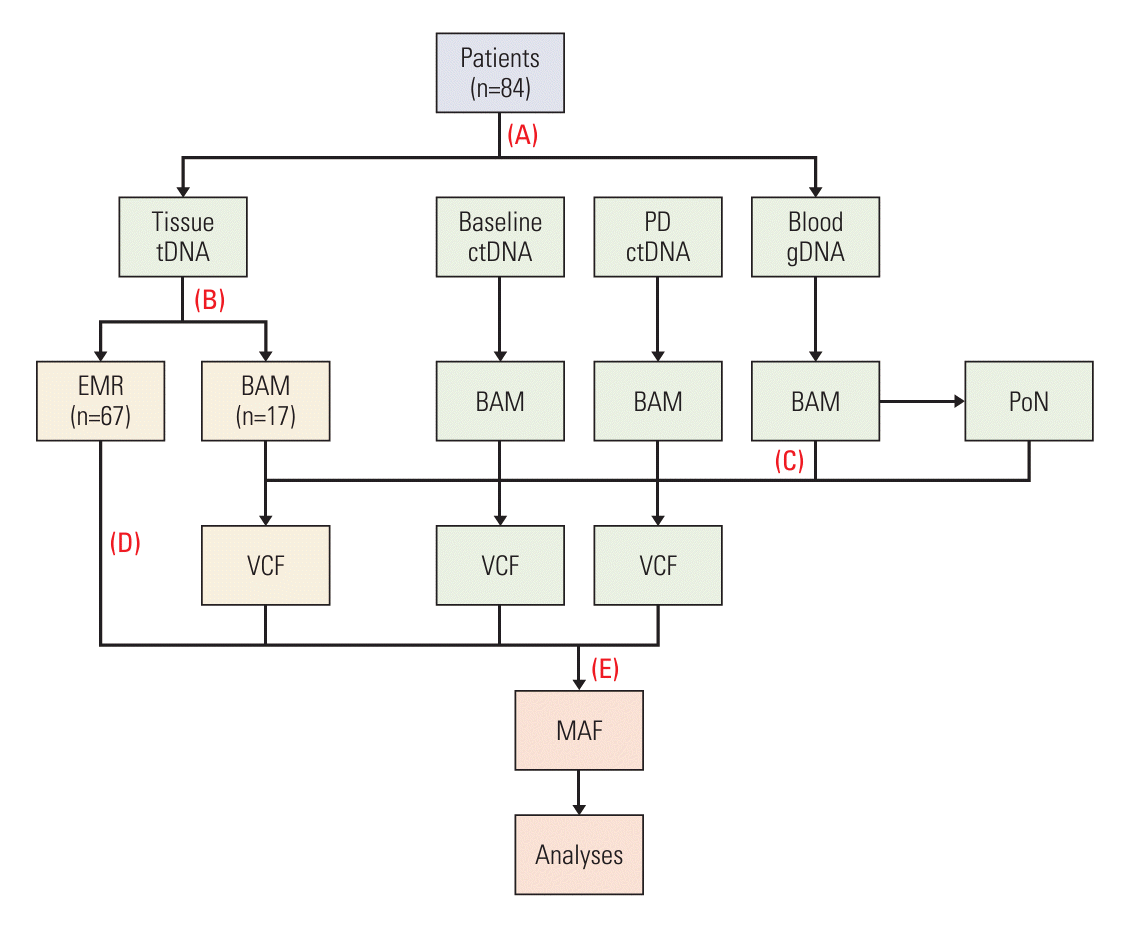
From targeted sequencing data, we performed somatic variant discovery for ttDNA, baseline-ctDNA, and PD-ctDNA (Fig. 1). To this end, we applied the Mutect2 command from GATK following GATK’s Best Practices workflow; for every tumor BAM file, we provided Mutect2 the matched normal (gDNA from blood) BAM file as well to call somatic mutations. Additionally, to further discriminate true somatic mutations from germline variants and potential artifacts from targeted sequencing, we provided Mutect2 a panel of normals, which was generated from blood BAM files using the CreateSomaticPanelOfNormals command from GATK, and also provided a large-scale collection of known germline variants from the Genome Aggregation Database (gnomAD) (downloaded on November 30, 2021) [17]. The output of Mutect2 was a variant call format (VCF) file for each tumor BAM file. Next, we used the FilterMutectCalls command from GATK to select only variants that have ‘PASS’ in the FILTER column of VCF. Filtered variants were functionally annotated using the online version of Ensembl Variant Effect Predictor (VEP) [18] with ‘RefSeq transcripts’ as the transcript database and the filtering option ‘Show one selected consequence per variant’. To focus our downstream analyses on significant driver mutations only, we only retained variants if they had ‘HIGH’ as the IMPACT rating from VEP or if they were a missense and either the PolyPhen-2 prediction was ‘probably_damaging’ or the SIFT prediction was ‘deleterious’ or the CLIN_SIG field was ‘pathogenic’. Of the 84 ttDNA samples, 67 represented EMR data from SNUBH cancer genomics testing. To automatically extract mutation information from EMR data, we devised a text parsing algorithm using the Python language. Parsed EMR data were then combined with VEP-annotated variants to create a multi-sample mutation annotation format (MAF) file using the pymaf submodule from the fuc package (https://sbslee-fuc.readthedocs.io/en/latest/api.html#module-fuc.api.pymaf) [19]. Lastly, mutations in the resulting MAF file were filtered out if they were not part of the 88 genes targeted by ACP1.
6. Visualization of the mutational landscape
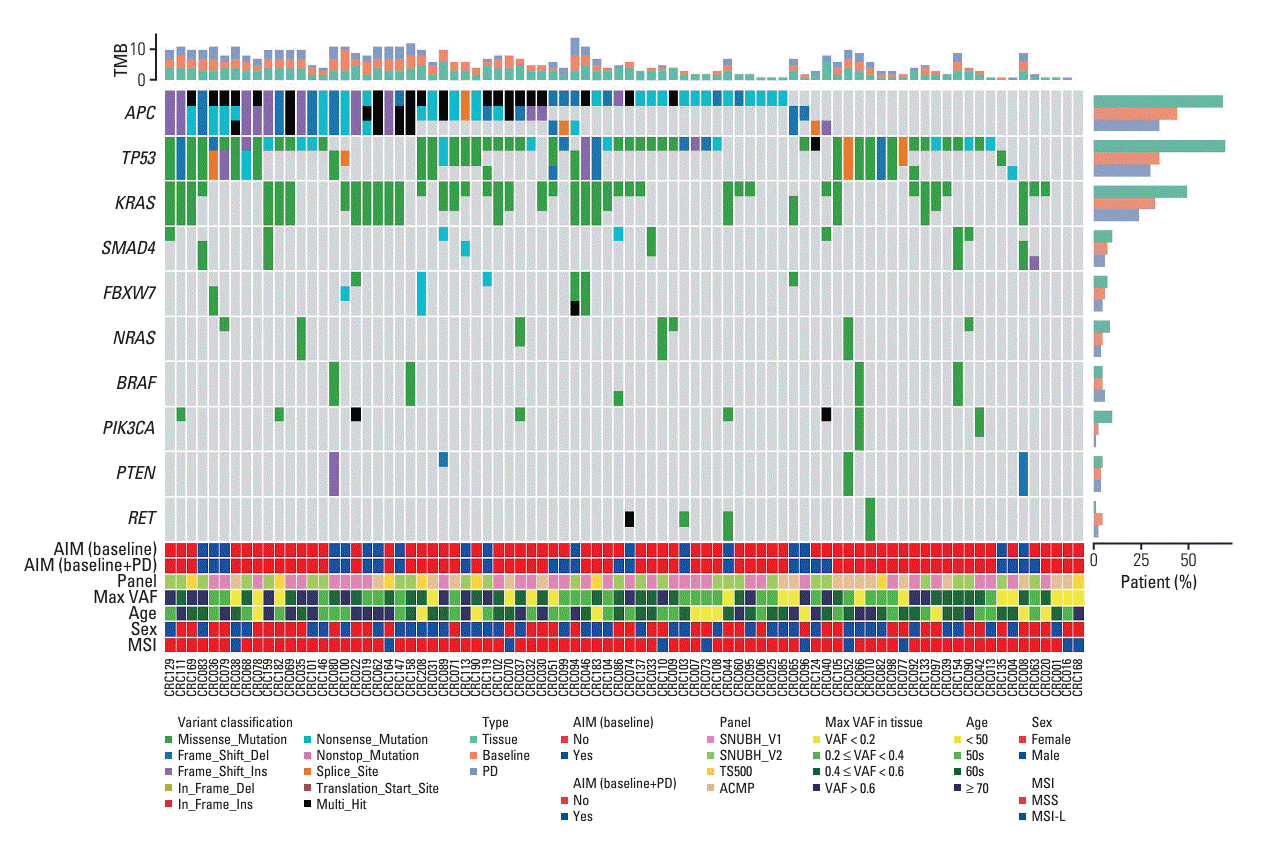
To visualize the mutational landscape of our samples, we created an oncoplot for the top ten genes with the highest mutation prevalence. To this end, we used several Python methods from the pymaf submodule including ‘pymaf.MafFrame.plot_waterfall_matched’, ‘pymaf.MafFrame.plot_tmb_ matched’, and ‘pymaf.MafFrame.plot_mutated_matched’. We also added to the oncoplot several clinical annotations including age, sex, and microsatellite instability (MSI) status to identify potential patient subgroups. Additionally, to show the frequency of specific amino acid changes for a given gene, we created a lollipop plot (also known as a stem plot) using the ‘pymaf.MafFrame.plot_lollipop’ method. Finally, we computed pairwise genotype concordance between the sample types using the ‘pymaf.MafFrame.get_gene_concordance’ method.
7. Correlation between DNA sample types
We performed pairwise comparisons between the sample types to assess their correlation in gene mutation prevalence and tumor mutational burden (TMB) using the Python methods ‘pymaf.MafFrame.plot_regplot_gene’ and ‘pymaf. MafFrame.plot_regplot_tmb’, respectively. Additionally, we compared the distribution of VAF between the sample types using the ‘pymaf.MafFrame.plot_vaf’ method.
8. Analysis of gene-gene interaction
A heatmap of negative log p-values indicating the strength of pairwise association was generated for the top ten genes with the highest mutation prevalence by the ‘pymaf.MafFrame.plot_interactions’ method. This method performed Fisher’s exact test to identify significant pairs of genes that are mutually exclusive or co-occurring. We manually validated our findings by creating a scatter plot for each gene pair using the ‘pymaf.MafFrame.plot_genepair’ method.
9. Assessment of cancer evolution dynamics
We visualized changes in relative VAF between the sample types using the ‘pymaf.MafFrame.plot_evolution’ method. This method sets the highest VAF in the sample as 100% and then subsequently normalizes other VAF values accordingly. For example, if the highest VAF for a given sample is 0.5 in the gene A and the gene B has a mutation of VAF=0.2, these numbers will be converted to 100% and 40%, respectively.
10. Identification of clonal versus subclonal mutations
We quantified the percentage of mutations that were either clonal or subclonal for the top ten genes with the highest mutation prevalence. A mutation was defined as clonal if the VAF was greater than or equal to 25% of the highest VAF in the sample; conversely, a mutation was defined as subclonal if the VAF was less than 25% [20]. To study the effect of changing the threshold, we also repeated the same analysis with 35%, 45%, and 55% as thresholds. The ‘pymaf.MafFrame.plot_clonality’ method was used for visualization.
Results
1. Patient characteristics and clinical outcomes
Between October 2015 and June 2021, a total of 208 patients with mCRC who underwent palliative first-line systemic anti-cancer treatment were consecutively enrolled. Among them, 84 patients with available tumor NGS results and baseline- and PD-ctDNA samples were analyzed in this study (Table 1). Of these, 72 tumor NGS results were generated from tumors obtained before treatment initiation, and the other 12 tumor NGS results were examined using tumor samples acquired after treatment initiation. The male-to-female ratio was 41:43. The median age was 59 years (range, 35 to 90 years). The objective response rate of palliative first-line treatment was 47.6% (S2 Table). During a median follow-up of 46.8 months (range, 15.2 to 83.1 months), the median PFS and OS were 12.2 months (95% confidence interval [CI], 9.8 to 14.6) (S3A Fig.) and 27.4 months (95% CI, 22.8 to 32.0) (S3B Fig.), respectively.
2. Summary of identified somatic mutations
Five different NGS assays were employed during the study period: ACP1, ACMP, SNUBH Pan-Cancer Panel version 1 (SNUBH_V1), SNUBH Pan-Cancer Panel ver. 2 (SNUBH_V2), and TruSight Oncology 500 (TS500). These panels targeted a total of 88, 558, 99, 559, and 523 cancer-related genes, respectively (S1 Table). All liquid specimens, including gDNA from blood, baseline-ctDNA, and PD-ctDNA, were sequenced using ACP1 (n=252). For ttDNA from tumor tissue, 17 patients without prior EMR sequencing data were newly sequenced with ACMP, while patients with existing EMR data were sequenced using either SNUBH_V1 (n=33), SNUBH_V2 (n=26), or TS500 (n=8). Because ACMP, SNUBH_V1, SNUBH_V2, and TS500 contained all 88 genes present in ACP1, we limited our analysis to those genes.
Excluding duplicated mutations, we identified a total of 202 driver mutations in 34 of the 88 genes. ttDNA had the highest occurrence of mutations (n=232), followed by baseline-ctDNA (n=155) and PD-ctDNA (n=117). These mutations were functionally annotated to be missense (n=272), nonsense (n=101), frameshift deletions (n=68), frameshift insertions (n=50), and splice sites (n=13). Of note, no mutations were found at all in a single patient (subject identifier: CRC168). In addition, no mutations were found in baseline-ctDNA and PD-ctDNA for 26.2% (22/84) and 39.3% (33/84) of the patients, respectively, while for ttDNA this percentage was only 4.8% (4/84).
3. Comparison of the mutational landscape of ctDNA and tumor tissue
To visualize the mutational landscape of our samples, we created an oncoplot for the ten most mutated genes (Fig. 2). These genes included APC, TP53, KRAS, SMAD4, FBXW7, NRAS, BRAF, PIK3CA, PTEN, and RET. The mutational prevalence generally decreased in the order of ttDNA, baseline-ctDNA, and PD-ctDNA. This pattern was particularly evident among the top three genes APC, TP53, and KRAS. For example, we observed mutations in APC from 67.9%, 44.0%, and 34.5% of the patients in ttDNA, baseline-ctDNA, and PD-ctDNA, respectively. Still, there were a considerable number of patients where mutations were only detected with ctDNA. For example, APC and KRAS mutations in the patient CRC065 were only detected with baseline- and PD-ctDNA. The same was true for SMAD4 in the patient CRC008 and for TP53 in the patient CRC080. When only the top ten genes were considered, 18 of the 84 patients (21.4%) were found to have additional mutations from sequencing baseline-ctDNA in addition to ttDNA alone. Moreover, since some mutations could emerge during treatment, the number of patients who were found to have additional mutations was increased to 25 (29.8%) when both baseline- and PD-ctDNA were considered.
We found high genotype concordance between ttDNA and ctDNA for the top ten genes (Table 2). Except for APC and TP53, the concordance between baseline- and PD-ctDNA was greater than 90%. The biggest difference between ttDNA and ctDNA was observed in APC and TP53 where the concordance ranged from 47.6 to 53.6%.
Different genes harbored different patterns of mutation frequency indicating distinctive mechanisms of action (e.g., gain- or loss-of-function). To highlight some of these patterns, lollipop plots for the top three genes APC, TP53, and KRAS are shown for each sample type (S4 Fig.). The majority (79 of 88) of KRAS mutations were found in codons 12 and 13 (e.g., p.G12D, p.G12V, p.G13D, etc.) which are well-characterized activating mutations. In comparison, the tumor suppressor genes APC and TP53 displayed a random distribution of mutations across the entire gene.
Expanding the oncoplot to include all 34 mutated genes, instead of just the top ten, revealed several additional features about the mutational landscape of our samples (S5 Fig.). First, we observed that the bottom nine genes were affected by singletons—i.e., for each gene, there was only one mutation from one of the three samples. These singletons were found in three, six, and zero samples from ttDNA, baseline-ctDNA, and PD-ctDNA, respectively. Second, the following eight genes had multiple mutations detected only with ctDNA: BRCA1, AXL, ERBB3, RB1, NTRK1, FLT3, EGFR, and CDH1. Moreover, except for FLT3 and CDH1, they each had one patient whose baseline- and PD-ctDNA detected the identical mutation. These observations increased the total number of patients who were found to have additional mutations from sequencing baseline-ctDNA, from 18 (21.4%, when only the top ten genes were considered) to 32 (38.1%, when all 34 mutated genes were included). When both baseline- and PD-ctDNA were considered, the number increased from 25 (29.8%, with the top ten genes) to 40 (47.6%, with all 34 mutated genes).
Compared with ttDNA and baseline-ctDNA, PD-ctDNA identified 13 novel mutations in 10 patients (11.9%). Among them, seven mutations in five patients (6.0%) were missense or nonsense mutations in tumor suppressor genes such as APC (CRC040, CRC051, and CRC094), TP53 (CRC004 and CRC051), SMAD4 (CRC063), and CDH1 (CRC040), which might implicate acquired resistance to anti-cancer drugs.
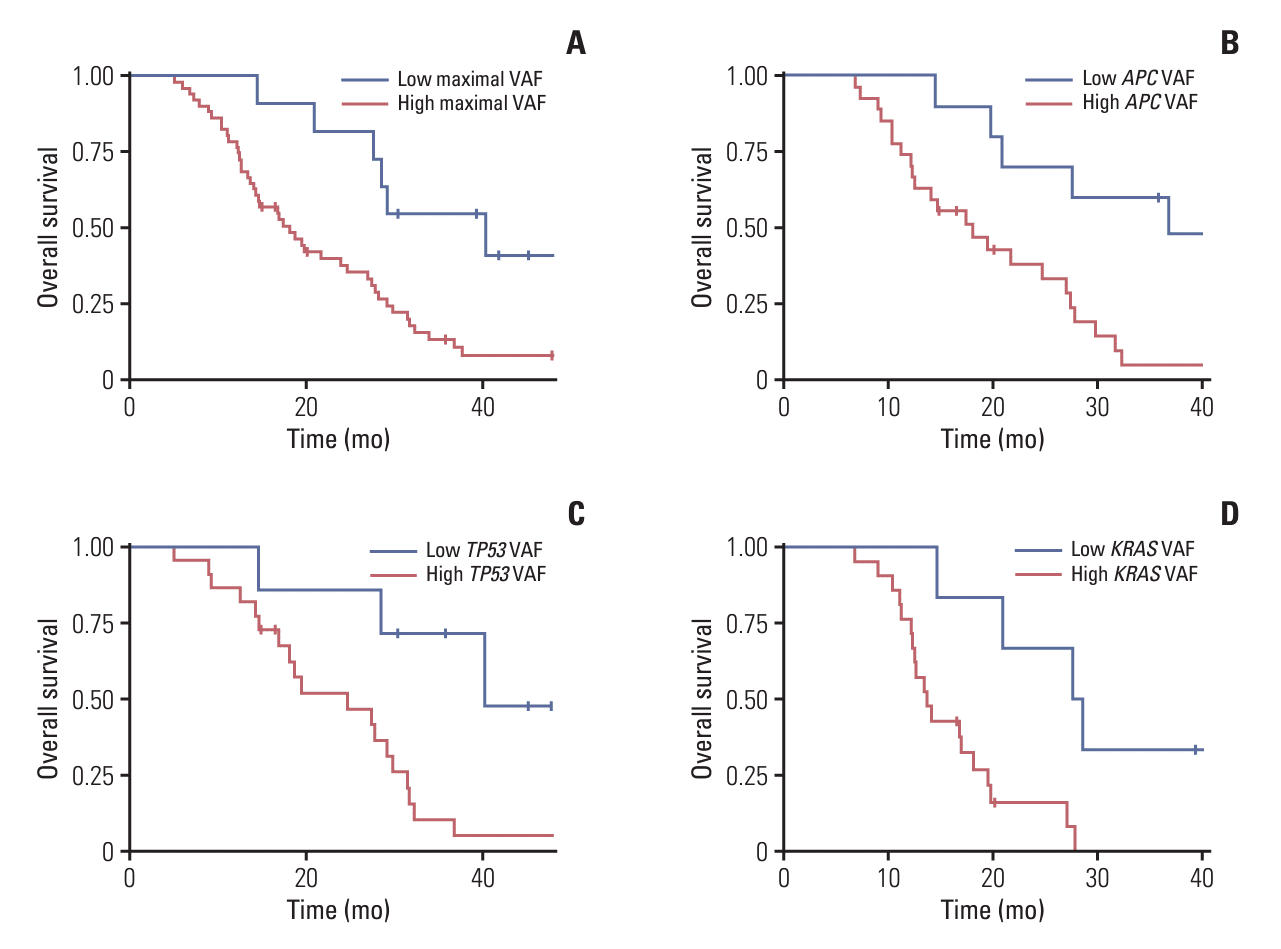
4. Survival outcomes according to VAF
Among patients who had any mutations in baseline-ctDNA (n=62), the maximal VAF was median 0.251 (range, 0.013 to 0.902). Patients with higher maximal VAF values of more than 0.059 showed significantly worse PFS (median, 10.5 months [95% CI, 9.0 to 12.0] vs. 17.1 months [95% CI, 14.6 to 19.7]; p=0.036) (S6A Fig.) and OS (median, 18.1 months [95% CI, 12.9 to 23.4] vs. 40.3 months [95% CI, 23.7 to 57.0]; p=0.010) (Fig. 3A). Moreover, among patients harboring APC, TP53, and KRAS mutations (n=37, 29, and 27, respectively), the median VAF values of APC, TP53, and KRAS mutations were 0.258 (range, 0.013 to 0.842), 0.377 (range, 0.021 to 0.902), and 0.194 (range, 0.037 to 0.680), respectively. The OS was significantly worse in those with high VAF values of APC (median, 18.1 months [95% CI, 11.0 to 25.3] vs. 36.8 months [95% CI, 18.2 to 55.4]; p=0.012) (Fig. 3B), TP53 (median, 24.7 months [95% CI, 12.6 to 36.8] vs. 40.3 months [95% CI, not calculated]; p=0.012) (Fig. 3C), and KRAS (median, 13.7 months [95% CI, 11.4 to 16.0] vs. 27.6 months [95% CI, 18.5 to 36.7]; p=0.005) (Fig. 3D) mutations when cutoff values of 0.133, 0.154, and 0.081 were applied, respectively.
The PFS of patients with APC mutation was significantly shorter in those with high APC VAF values (median, 10.8 months [95% CI, 8.2 to 13.3] vs. 16.9 months [95% CI, 13.4 to 20.5]; p=0.019) (S6B Fig.). Similarly, according to the VAF values of TP53 and KRAS mutations, the PFS tended to be different without statistical significance (p=0.120 and p=0.114, respectively) (S6C and S6D Fig.).
5. Pairwise correlation results
We performed pairwise comparisons between the sample types to assess potential correlation in various metrics. The mutational prevalence of the top ten genes in baseline-ctDNA was well correlated with the mutational prevalence in ttDNA (R2 =0.96, p < 0.001) (S7A Fig.). In all three comparisons, the top four genes were APC, TP53, KRAS, and SMAD4. The same patterns were observed for PD-ctDNA vs. ttDNA (R2 =0.97, p < 0.001) as well as the baseline- vs. PDctDNA (R2 =0.99, p < 0.001). In contrast, the TMB in baseline-ctDNA only showed a weak association with the TMB in ttDNA (R2 =0.24, p < 0.001) (S7B Fig.). The association between PD-ctDNA and ttDNA was even weaker (R2 =0.16, p < 0.001). Interestingly, the TMB in baseline-ctDNA and that of PDctDNA showed a relatively strong correlation (R2 =0.40, p < 0.001). Next, we compared the distribution of VAF between the sample types for the top ten genes (S7C Fig.). In almost all cases, VAF decreased in the order of ttDNA, baseline-ctDNA, and PD-ctDNA.
6. Discovery of gene-gene interactions
To unearth any possible relationships (i.e., co-occurring and mutually exclusive) between the mutated genes, we created a heatmap of statistical significance using Fisher’s exact test for the top ten genes (S8A Fig.). When using p < 0.05 as the significance cutoff, four gene pairs (KRAS-TP53, PIK3CA-BRAF, PTEN-NRAS, and PTEN-BRAF) were co-occurring, while three gene pairs (KRAS-BRAF, KRAS-NRAS, and APC-RET) were mutually exclusive. Four gene pairs involving KRAS were visualized: KRAS-APC, KRAS-TP53, KRAS-BRAF, and KRAS-NRAS (S8B-S8E Fig.). In addition, the rest of the pairs also showed evidence of gene-gene interactions (S9 Fig.).
7. Detection of clonal and subclonal mutations
We assessed the clonal vs. subclonal landscape of mutations in our patient cohort. According to previous literature [20], we defined a mutation as clonal if the VAF was equal to or above 25% of the highest VAF in the sample and as subclonal if it was less than this cutoff. We found that only 4.0% (20/504) of the detected mutations were subclonal. To study the effect of different thresholds, we repeated the same analysis with 35%, 45%, and 55% as thresholds (S10 Fig.). Among the ten genes with the highest mutational prevalence, the three genes most likely to be subclonal were PIK3CA, PTEN, and RET.
8. Individual variation in cancer evolution dynamics
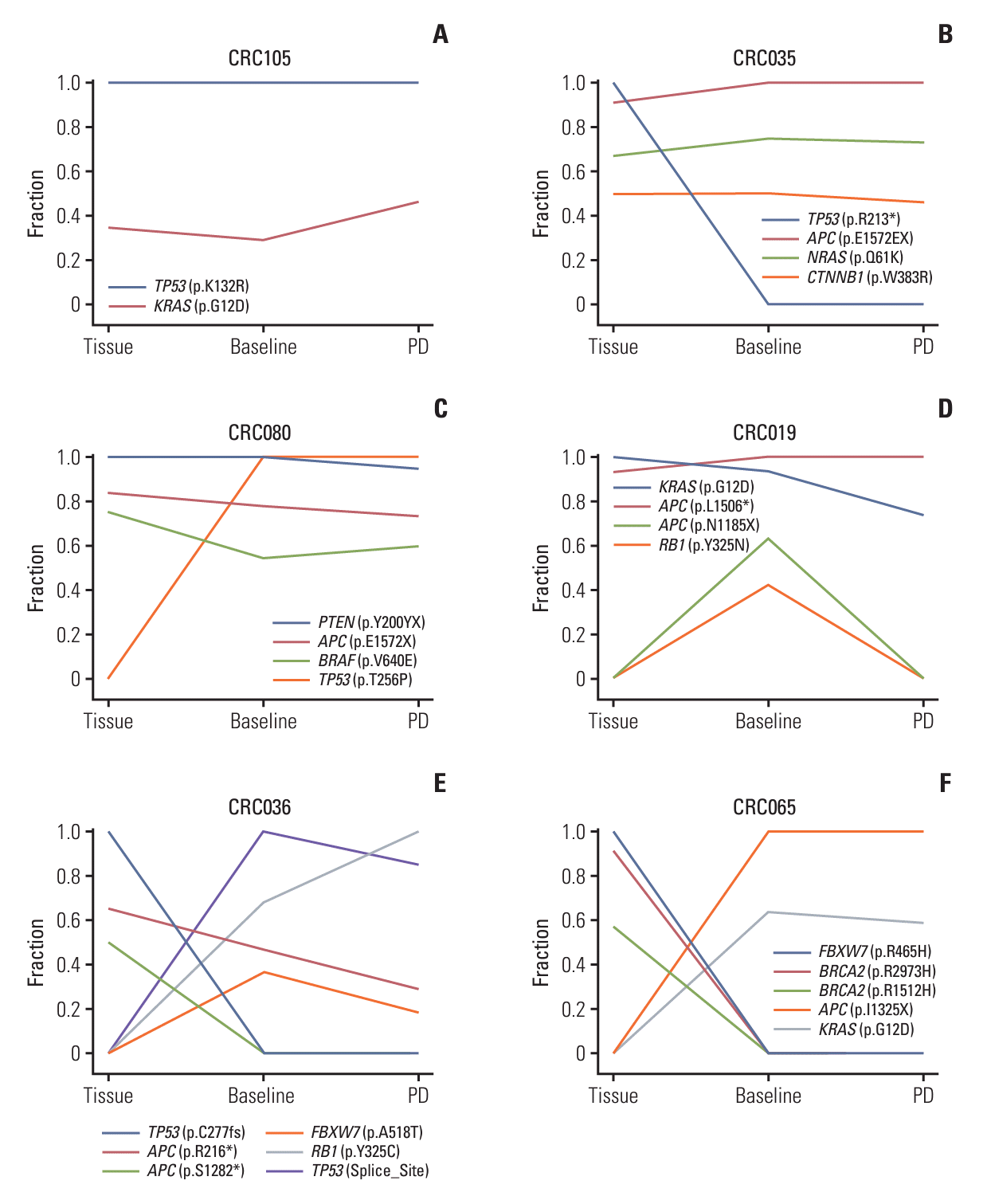
To assess the dynamics of clonal evolution, we visualized changes in normalized VAF across the different sample types in six representative patients (Fig. 4). In the first patient CRC105 (Fig. 4A), the TP53 p.K132R mutation presented the highest VAF in all the samples and thus was assigned 100%. Subsequently, the VAF of the other mutation KRAS p.G12D was normalized compared to the TP53 mutation in each sample, revealing that the two mutations were maintained stably during treatment. In the second patient CRC035 (Fig. 4B), all four of the observed mutations were stably maintained except TP53 p.R213* which was detected in tissue with the highest VAF, but not at all in ctDNA. The third patient CRC080 (Fig. 4C) showed an opposite pattern where all four of the observed mutations were stably maintained except TP53 p.T256P which was detected in ctDNA with high VAF levels (99.7% and 100% in baseline- and PD-ctDNA, respectively), but none in tissue. In the fourth patient CRC019 (Fig. 4D), the two mutations APC p.L1506* and KRAS p.G12D were consistently observed across the samples, but the other two mutations APC p.N1185X and RB1 p.Y325N were only observed in baseline-ctDNA. The fifth patient CRC036 (Fig. 4E) produced a complex trajectory of cancer evolution where there was only one common mutation, APC p.R216*, between tissue and ctDNA.
In the last patient CRC065 (Fig. 4F), there were no common mutations between ttDNA and ctDNA, which is suggestive of the presence of two independent cancers. The CRC065 patient was diagnosed with stage I rectal cancer nine years before developing liver metastasis. At the time of liver metastasis, a rectal mass developed again at the previous anastomosis site. However, because of limited tissue availability, the ttDNA analysis was conducted using the old rectal cancer specimen from nine years ago. Therefore, we can conclude that the second rectal mass did not originate from the previous one but was a newly developed metachronous cancer.
Discussion
This study compared clinical tumor NGS data and UMI-based baseline- and PD-ctDNA data from 84 patients with mCRC receiving palliative first-line systemic treatment. While ttDNA analysis exhibited higher sensitivity than ctDNA analysis, the latter demonstrated validity and potential value when ttDNA was not available. UMI technology, in particular, enhanced sensitivity and reliability in our cancer genome profiling, enabling the detection of rare mutations— as low as a VAF of 0.36%—that would have otherwise gone unnoticed without error correction using UMI deduplication. Moreover, repetitive ctDNA analysis at PD identified novel somatic mutations during treatment.
In our data, 32 patients (38.1%) were found to have additional mutations from baseline-ctDNA analysis in addition to ttDNA alone. This number increased to 40 patients (47.6%) when both baseline- and PD-ctDNA results were analyzed. PD-ctDNA identified 13 novel mutations in 10 patients (11.9%), including seven missense or nonsense mutations in tumor suppressor genes in five patients (6.0%), possibly linked to clonal evolution and anti-cancer drug resistance. High VAF values in baseline-ctDNA correlated with worse survival outcomes.
Although ctDNA analysis cannot entirely replace ttDNA analysis when tumor tissue is available, our data indicated substantial similarity between ctDNA-based and ttDNA-based profiling, showing potential usefulness when ttDNA is inaccessible. While mutational prevalences of top genes aligned well between ttDNA and baseline-ctDNA, TMB showed relatively weaker correlation possibly due to panel size limitations in our ctDNA platform.
Patients with high maximal VAF values showed significantly worse PFS and OS. This trend was consistent for OS in individuals with high VAF values of APC, TP53, and KRAS mutations. Conventional imaging techniques struggle to quantify cancer patients’ overall disease burden accurately. Although multiple factors determine the VAF values, it is known that baseline-ctDNA VAF values may mirror the overall tumor burden [21]. Previous studies have highlighted the prognostic significance of VAF values in cancer patients [22,23]. Our data further support baseline-ctDNA analysis as a relatively straightforward method to approximate overall tumor burden and predict prognosis. However, VAF values can be notably influenced by locus loss-of-heterozygosity [24]. Therefore, combining both measures—VAF and loss-of-heterozygosity, if applicable, would offer a more comprehensive assessment.
Our ctDNA analysis unveiled insights relevant to CRC biology. The observed co-occurrence of KRAS and TP53 mutations aligns with the stepwise carcinogenesis of CRC sequence of APC > KRAS > SMAD4 > TP53 [25]. Similarly, the mutual exclusiveness of KRAS-BRAF and KRAS-NRAS is not surprising, given that KRAS, BRAF, and NRAS are key oncogenes in the Ras > Raf > MEK > MAP-kinase signaling pathway and tend to be mutually exclusive [26]. Consistent with a previous pan-cancer study [27], genes with higher mutational prevalence were more likely to exhibit clonal mutations, while those with lower mutational prevalence were more inclined towards subclonal mutations.
In certain patients with available tumor NGS results, ctDNA analyses may provide an added benefit in identifying novel mutations. In addition, a substantial proportion of patients exhibited different mutational profiles before and after treatment, emphasizing the role of ctDNA analysis in monitoring clonal evolution in cancer. Prior studies have found that EGFR inhibition induces gain-of-function mutations in known oncogenes like KRAS, NRAS, BRAF, MET, and EGFR in mCRC patients [28-31]. In our study, our ctDNA platform discovered seven novel loss-of-function mutations in tumor suppressor genes in five patients at PD, but no new activating mutation in oncogenes. Only one of these five patients received the anti-EGFR monoclonal antibody, cetuximab, while the remaining four received the anti-VEGF antibody, bevacizumab. Hence, it is speculated that acquired mutations during treatment might not solely be induced by EGFR inhibitors but also by other anti-cancer drugs, affecting not only oncogenes but also tumor suppressor genes. It is also possible that the acquired resistance of EGFR inhibitors is driven by undiscovered mechanisms of tumor suppressor genes or epigenetic changes that cannot be detected by ctDNA analysis.
Interestingly, our ctDNA study aided in rectifying the diagnosis of metachronous malignancy in patient CRC06, a distinction that might have been challenging to discern from locoregional recurrence using conventional pathological testing methods. This finding was an incidental discovery during our study, highlighting the potential of the ctDNA platform in diagnosing complex cases solely through blood sampling.
A limitation of this study arose from our intent to utilize real-world clinical NGS data on ttDNA, which used a variety of target panels for ttDNA. Most patients had already undergone testing with three different panels (SNUBH_V1, SNUBH_V2, and TS500) as part of routine clinical practice using ttDNA, while the remaining 17 patients underwent ACMP ttDNA analysis for the sole purpose of conducting this study. Nevertheless, it is noteworthy that all 88 cancer-associated genes included in the ACP1 ctDNA panel were consistently covered across the four ttDNA NGS panels. Furthermore, no distinct patient subgroups were identified within the mutational landscape based on factors such as max VAF, age, sex, targeted panel, and MSI status. In addition, the relatively low concordance of ctDNA and ttDNA in APC and TP53 mutations could be attributed to the difference in the target probe design of the ACP1 panel. The limited sample size and heterogeneous characteristics of our patient cohort may potentially impede the validity of conducting survival analysis related to VAF values. For instance, although somatic mutations in KRAS, NRAS, BRAF, and ERBB2 genes are known prognostic factors in the targeted therapy era [4-7], the small sample size of our study limited us from determining whether these mutations act as prognostic factors in survival analysis.
In conclusion, although ttDNA retains higher sensitivity compared to ctDNA, our UMI-based ctDNA platform demonstrated validity and substantial value, especially when ttDNA was unavailable. The integration of ttDNA, baseline-ctDNA, and PD-ctDNA analyses provided supplementary insights, uncovering previously undetected pathogenic mutations and illustrating clonal dynamics during treatment. Additionally, baseline-ctDNA’s VAF values predicted prognosis after treatment. This approach holds promise in precisely deciphering the cancer genome and guiding tailored therapy by identifying unique resistance mechanisms in each patient, thereby shaping the future trajectory of clinical oncology.
 XML Download
XML Download