

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Oxidative stress is a recognized consequence of an imbalance between free radical generation and the body’s antioxidant defenses, ultimately leading to cellular and tissue damage [1]. This imbalance has been implicated in the development and progression of various diseases, including cardiovascular disease, neurodegenerative disease, and cancers. Therefore, the identification of novel antioxidant substances is imperative for advancing therapeutic strategies aimed at mitigating these health issues [2,3]. Traditionally, the assessment of antioxidant capacity has relied on in vitro biochemical assays, such as the 2,2-diphenyl-1-picrylhydrazyl (DPPH) and 2,2'-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) tests [4,5]. While the DPPH and ABTS assays are effective, they are often labor-intensive and time-consuming, requiring substantial amounts of sample material. Moreover, the complexity of these methods limits their scalability and efficiency and hinders the high-throughput screening of compounds, particularly in early-stage drug discovery processes.
The emergence of machine learning (ML) technologies offers promising alternative approaches to enhance the efficiency of antioxidant identification [6,7]. ML has the potential to overcome the limitations of traditional methods by facilitating rapid, cost-effective in silico screening of vast chemical libraries for antioxidant activity [8,9]. By leveraging accumulated histological data sets of chemical bioassays and advanced algorithmic models, ML can predict the antioxidant potential of compounds solely based on their chemical structures. This capability expedites the discovery process and enriches our understanding of structure-activity relationships within antioxidant compounds [10].
This study investigated the applicability of various ML algorithms for predicting the antioxidant activity of chemical compounds. We curated a dataset of chemical structures annotated with experimentally determined antioxidant activities and employed well-established ML algorithms to develop predictive models. We demonstrated the efficacy of these models in predicting antioxidant activities and assessed the reliability of prediction as a preliminary screening tool before extensive in vitro validation.
METHODS
Acquisition of antioxidant activity data
Publicly available antioxidant activity data for a diverse range of compounds was retrieved from the PubChem database. To control methodological consistency within the dataset, data was exclusively sourced from well-established assays: ABTS and DPPH. ABTS and DPPH assays quantify a compound's free radical scavenging (ABTS) or reduction (DPPH) capacity, reflected by a measurable color change [4,5]. Selection of these assays was based on their widespread adoption in rapid antioxidant potential screening due to their simplicity and effectiveness. PubChem searches employing the keywords "DPPH" and "ABTS" identified a total of 1,651 DPPH and 366 ABTS assays encompassing 19,454 compounds.
Data preprocessing
To eliminate redundancy arising from compounds with identical structures but varying identifiers, the International Union of Pure and Applied Chemistry (IUPAC) InChIKeys was adopted as the unique identifier. The RDKit Python package was utilized to convert the Simplified Molecular Input Line Entry System (SMILES) strings for each compound into IUPAC InChIKeys. This process refined the initial 19,454 PubChem CIDs into 10,053 unique compounds. Subsequently, Extended-Connectivity Fingerprints (ECFP) with a radius of 4 (ECFP-4) were generated using the RDKit Python package to represent chemical structures based on each compound’s SMILES string. Compounds were categorized into four activity groups for each assay: ‘Active’, ‘Inactive’, ‘Unspecified’, or ‘Inconclusive’, and those with solely ‘Inconclusive’ and ‘Unspecified’ designations across all assays were excluded. In addition, compounds exhibiting inconsistent activity results between the two assays were eliminated. To enhance the dataset's comprehensiveness, 24 well-known antioxidant compounds documented in prior studies were incorporated and designated as the ‘Active’ set [11].
Structural similarity calculation
The Tanimoto coefficient [12] between each pair of all compounds was computed using their ECFP-4 fingerprint via the DataStructs Python package. ECFP-4 fingerprint is a binary vector of 2,048 bits in length, where each bit represents the presence (set to 1) or absence (set to 0) of specific circular substructures within a molecule. The Tanimoto coefficient itself is a value between 0 and 1, representing the degree of structural similarity between two molecules. A higher coefficient indicates a greater degree of structural similarity. The calculation is as follows:
where Nab is the number of common features between the two compounds, Na is the total number of features in compound A, and Nb is the total number of features in compound B. To visualize the relationships between the compounds based on the calculated Tanimoto coefficients, a compound-compound network was constructed. In this network, each compound is represented by a node, and edges connect nodes that exhibit a Tanimoto coefficient exceeding a predefined threshold (set to 0.7 in this case). Cytoscape 3 (version 10.3.1), an open-source software platform for network visualization, was used to generate this network representation.
ML model training and evaluation
Five ML algorithms were chosen for their capability to model complex relationships between molecular structure and antioxidant activity: Support Vector Machine (SVM), Logistic Regression (LR), XGBoost (XGB), Random Forest (RF), and Deep Neural Network (DNN). Each model was trained via functions from the scikit-learn Python package with default parameter settings to prevent overfitting and ensure generalizability (Table 1, Supplementary Fig. 1). For robust and unbiased evaluation, five-fold cross-validation was conducted using two data splitting strategies: (i) random splitting and (ii) scaffold splitting. To achieve scaffold splitting, all compounds were classified into scaffold groups using a Scaffold Network Generator [13] implemented in the RDKit Python package. This tool organized compounds into a hierarchical tree structure, with groups ranging from single-ring to a maximum of 15-ring structures. Consequently, 1,931 compounds were categorized into 778 scaffold groups. The training data was then split based on scaffold membership, ensuring the testing set contained compounds with unseen scaffold structures. Using both splitting strategies, the five-fold cross-validation was repeated 100 times. Performance metrics including accuracy, precision, recall, F1-score, and area under the receiver operating characteristic curve (AUROC) were computed for each iteration across all five models using the scikit-learn Python package.
Table 1
Machine learning models and hyperparameter settings
![]()
RESULTS
Overall analytic process
This study evaluated well-established ML algorithms for predicting the antioxidant activity of compounds based solely on their molecular structure represented by SMILES strings (Fig. 1). A dataset of 19,454 compounds with antioxidant activity data and corresponding SMILES information was collected from PubChem and literature searches. Following a rigorous data-cleaning process, a final set of 1,931 compounds (1,092 active, 839 inactive) was used to develop antioxidant activity prediction models. The ECFP-4 fingerprints, encoding the chemical environment of each molecule, were used as input features for the five ML algorithms (SVM, LR, XGB, RF, and DNN). A cross-validation scheme was implemented to identify the most suitable model for predicting antioxidant activity. The generalizability of the chosen model was further evaluated using external datasets of natural product compounds.
Fig. 1
Schematic illustration of the analytic workflow for constructing antioxidant compound prediction models employing five ML algorithms.
ML, machine learning; ABTS, 2,2’-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid); DPPH, 2,2-diphenyl-1-picrylhydrazyl; SMILES, Simplified Molecular Input Line Entry System; ECFP-4, Extended-Connectivity Fingerprints with a radius of 4; RF, Random Forest; SVM, Support Vector Machine; XGB, XGBoost; LR, Logistic Regression; DNN, Deep Neural Network.
![]()
Structural diversity of compounds and its relationship to antioxidant activity
To quantify the chemical diversity within the dataset and explore potential structural relationships associated with antioxidant activities, pairwise Tanimoto similarity coefficients [12] were calculated for all compounds. A compound-compound network, where nodes represented individual compounds, with edges connecting nodes that shared a Tanimoto coefficient exceeding a threshold of 0.7, was constructed to explore structurally similar compounds in the collected compounds (Fig. 2A). This network revealed that active and inactive compounds formed separate clusters, suggesting a positive correlation between structural similarity and antioxidant activity. However, an intriguing exception was observed within a cluster enriched with flavonoids structurally related to chrysin, a potent antioxidant flavone (Fig. 2B). While all compounds within this cluster shared a flavone backbone similar to the potent antioxidant chrysin, their antioxidant capacities diverged. This highlights the importance of the specific arrangement of functional groups within the flavone structure. Chrysin, with hydroxyl groups at the 5 and 7 positions of the A ring, exhibits strong antioxidant activity due to the well-documented radical scavenging properties of these groups [14]. The addition of hydroxyl group at the 3' position of the B ring (as in apigenin) or at both the 3' and 4' positions (as in luteolin) enhances antioxidative activity. However, the addition of a hydroxyl group at the 2' position of the B ring (as in 5,7,2'-trihydroxyflavone) does not result in antioxidative effects. Substitution of hydroxyl groups with methyl groups diminishes activity; for example, tectochrysin and acacetin (methylated at the 7 position of the A ring and 3' position of the B ring, respectively) and apigenin 7,4'-dimethyl ether (methylated at the 7 position of the A ring and 4' position of the B ring) both lose their antioxidative properties. These observations highlight that the hydroxyl groups at the 5 and 7 positions on the A ring, with an unblocked 3' hydroxyl group on the B ring, are crucial for the antioxidant activity of flavones. This data suggests that while overall structural features are informative in determining antioxidant activity, it is the precise arrangement of functional groups within the molecule that ultimately dictates its efficacy.
Fig. 2
Compound structural diversity and its association with antioxidant activity.
(A) Network visualization of compound relationships, where nodes represent individual compounds and edges connecting the nodes indicate high structural similarity between compounds (Tanimoto coefficient exceeding 0.7). Nodes are colored according to their experimentally determined antioxidant activity (gray for inactive, red for active). The network is segregated into three sub-panels: (i) a network containing only interconnected active compounds, (ii) a network with solely interconnected inactive compounds, and (iii) a network with a mixture of active and inactive interconnected compounds. (B) A representative example of a network module containing both active and inactive compounds. This highlights the potential for structurally similar compounds to exhibit diverse antioxidant properties. The border color of each node corresponds to its experimentally determined antioxidant activity (gray for inactive, red for active).
![]()
Model performance and evaluation
Five ML models, including SVM, LR, XGB, RF, and DNN, were employed to learn these subtle yet important structural features for predicting antioxidant activity based on ECFP-4 fingerprints. Model performance was evaluated using two five-fold cross-validation (5-fold CV) schemes: random splitting and scaffold splitting (Fig. 3). Random splitting divided the training data into five equal folds, where each fold is used for testing once while the remaining four are used for training. On the other hand, scaffold splitting grouped structurally similar compounds together in each fold. This approach evaluates the model's ability to predict the activity of compounds with novel scaffold structures not present in the training data, a crucial capability for discovering novel antioxidants with distinct chemical backbones (known as scaffold hopping).
Fig. 3
Performance comparison of five ML models.
(A, B) Representative receiver operating characteristic (ROC) curves and precision-recall curves were obtained from a single iteration of 5-fold cross-validation with (A) random splitting and (B) scaffold splitting. ML, machine learning; SVM, Support Vector Machine; LR, Logistic Regression; XGB, XGBoost; RF, Random Forest; DNN, Deep Neural Network.
![]()
All models achieved commendable performance on the random splitting CV (Fig. 3A and Table 2). RF outperformed other models in all metrics, including accuracy (0.908 ± 0.004), precision (0.912 ± 0.004), recall (0.927 ± 0.005), F1 score (0.919 ± 0.003), and AUROC (0.968 ± 0.002). SVM and XGB showed competitive performance with accuracies exceeding 0.900 and AUROC above 0.955. LR performed similarly but with slightly lower recall and F1 scores. DNN, while achieving a respectable accuracy (0.877 ± 0.015), exhibited higher variability in its metrics, suggesting potential overfitting or sensitivity to the composition of training data.
Table 2
Model performance obtained from 5-fold CV with random-splitting
Performance metrics were obtained across 100 iterations of 5-fold CV with random-splitting (average ± standard deviation). CV, cross-validation; SVM, Support Vector Machine; LR, Logistic Regression; XGB, XGBoost; RF, Random Forest; DNN, Deep Neural Network, AUROC, area under the receiver operating characteristic curve.
![]()
Notably, all models maintained good performance on scaffold-splitting CVs, albeit their scores were slightly lower compared to random-splitting (Fig. 3B and Table 3). All models except DNN maintained high accuracies above 0.900, with SVM and XGB recording the highest (0.906 ± 0.007). Precision was notably higher for LR (over 0.918), while RF and SVM demonstrated superior recall rates (both exceeding 0.950). F1 scores remained consistent and high for SVM, LR, XGB, and RF. However, DNN's performance significantly declined, suggesting its lower robustness to scaffold-based splits. Similarly, AUROC scores for SVM, LR, XGB, and RF remained high, demonstrating their ability to distinguish classes across diverse data segmentation.
Table 3
Model performance from 5-fold CV with scaffold-splitting
Performance metrics were obtained across 100 iterations of 5-fold CV with scaffold-splitting (average ± standard deviation). CV, cross-validation; SVM, Support Vector Machine; LR, Logistic Regression; XGB, XGBoost; RF, Random Forest; DNN, Deep Neural Network, AUROC, area under the receiver operating characteristic curve.
![]()
Overall, the results suggest that SVM and RF are well-suited for predicting antioxidant activity based on ECFP-4 fingerprints, exhibiting both high accuracy and generalizability across splitting methodologies.
Discriminative power of RF and SVM on the antioxidant activity of structurally similar compounds
To assess the ability of RF and SVM models to capture subtle structural features that are important for predicting antioxidant activity, we investigated their performance in differentiating between active and inactive compounds with high structural similarity. We focused on three network modules, each containing reference active compounds (chrysin, eriophorin A, and 4-(1H-indol-2-yl)aniline) (Fig. 4). Within the chrysin module (Fig. 2B), both models accurately classified chrysin and luteolin as active and apigenin as inactive, despite their high Tanimoto coefficient (> 0.7) (Fig. 4A). However, their performance diverged in other modules. Specifically, the RF model exhibited superior discriminatory power in the eriophorin A module (Fig. 4B), correctly classifying eriophorin A as active and eriophorin B and cajanin as inactive, even with their high structural similarity. Conversely, the SVM model, while differentiating active from inactive compounds, underestimated eriophorin A's activity, classifying it as inactive. A similar trend emerged in the 4-(1H-indol-2-yl)aniline module (Fig. 4C). The RF model accurately predicted active compounds, while the SVM model struggled. These findings demonstrate that, while both models can discriminate between some highly similar compounds, the RF model exhibits a stronger ability to distinguish active and inactive compounds with a high degree of structural similarity across diverse scaffold compounds. This suggests that RF models may be better suited to capture subtle structural variations that significantly influence antioxidant activity.
Fig. 4
Comparison of predicted antioxidant activity scores for structurally similar compounds.
(A) Predicted activity scores of compounds within the chrysin network module, as determined by RF and SVM models. (B, C) Network modules for (B) eriophorin A and (C) 4-(1H-indol-2-yl)aniline. The left panel shows the network where nodes represent compounds and edges represent high structural similarity (Tanimoto coefficient exceeding 0.7). The right panel displays the corresponding predicted activity scores obtained by the RF and SVM models for each compound within the respective network module. The border color of each node indicates its experimentally determined antioxidant activity (gray for inactive, red for active). RF, Random Forest; SVM, Support Vector Machine.
![]()
Prediction of antioxidant activity of natural products
To evaluate the generalizability of SVM and RF models, we performed external validation using a dataset of natural product compounds from the BATMAN (Bioinformatics Analysis Tool for Molecular mechANism of Traditional Chinese Medicine) database [15]. BATMAN offers a comprehensive resource for bioactive compounds found in traditional medicine and other natural products. We retrieved chemical structure data for 1,708 well-defined ingredient compounds extracted from 8,404 medicinal plants. A subset of 1,594 compounds not included in the training set was selected for unbiased evaluation. These natural compounds were then subjected to the SVM and RF models to predict their potential antioxidant activity (Supplementary Table 1). Subsequently, the top candidates with the highest average scores obtained from both models were shortlisted for further investigation. Notably, this shortlist was significantly enriched for highly hydroxylated flavonoids (Table 4). These specific classes of compounds are recognized for exhibiting antioxidant activity through free radical scavenging, metal chelation, and involvement in redox reactions [16-23].
Table 4
Top ten predicted antioxidant compounds from the BATMAN database
| Compound | PubChem CID | Structure | SVM | RF | No. of reference | Reference reporting antioxidant activity |
|---|---|---|---|---|---|---|
| Quercetin hydrate | 16212154 |
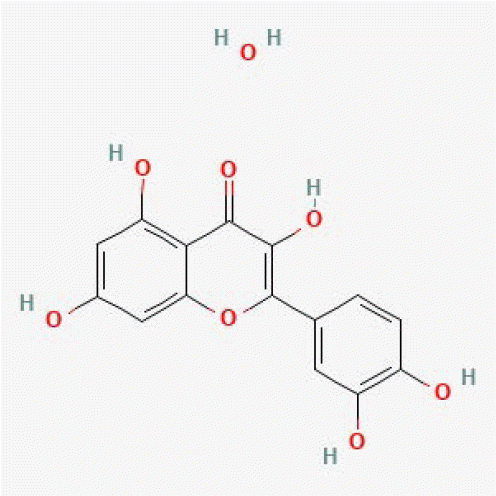
|
1.00 | 1.00 | 29 | [16] |
| Cyanidin chloride | 68247 |
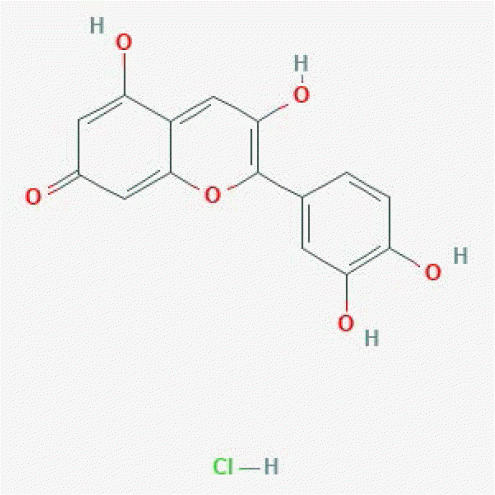
|
0.99 | 0.99 | 3,860 | [17] |
| Quercetagetin | 5281680 |
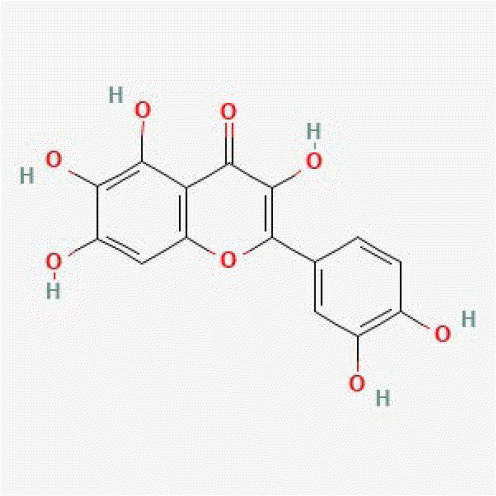
|
1.00 | 0.99 | 679 | [18] |
| Quercetin 3-O-rhamnoside | 5353915 |
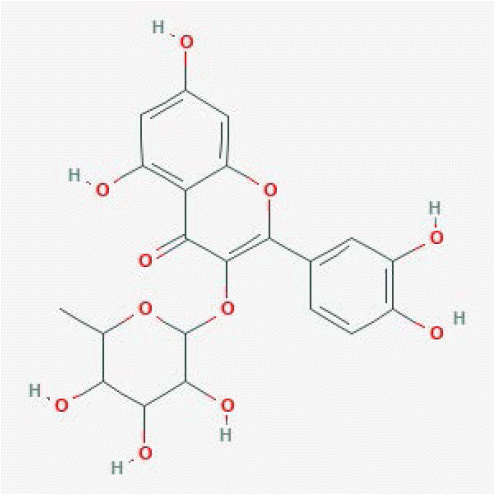
|
0.99 | 1 | 6 | [19] |
| Hispolon | 10082188 |
|
0.99 | 1.00 | 86 | [20] |
| Avicularin | 5490064 |
|
0.98 | 1 | 1,056 | [21] |
| Isoquercitrin | 51402807 |
|
0.98 | 1 | 2,246 | [22] |
| Delphinidin chloride | 128853 |
|
1.00 | 0.99 | 1,003 | [23] |
| Delphinidin | 68245 |
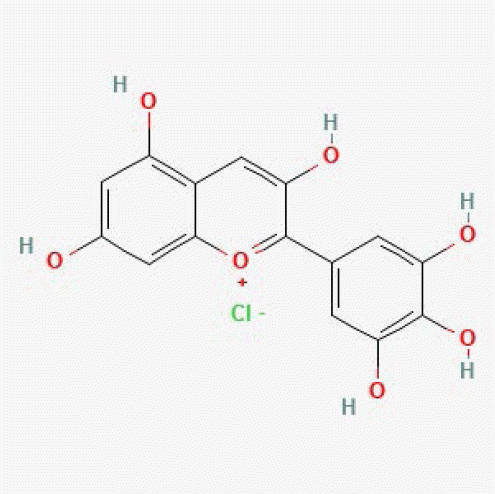
|
1.00 | 0.99 | 2,624 | [17] |
| Ellagic acid dihydrate | 16760409 |
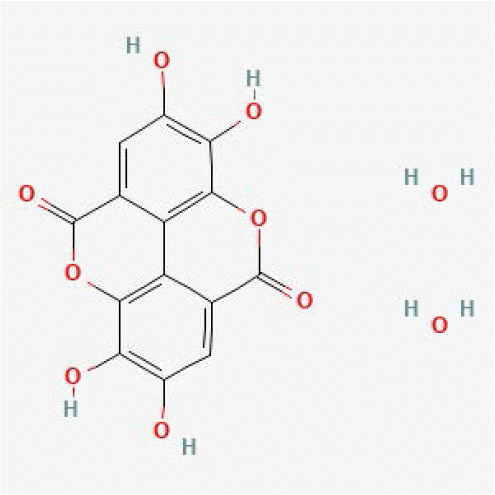
|
0.99 | 0.99 | 15 | - |
![]()
To explore natural compounds with novel antioxidant bioactivities, a literature search was conducted using SciFinder [24]. This search yielded publication counts for each compound, providing an indicator of their prior investigation in the context of biological activity. Most of the top 10 compounds are well-studied (median publication count = 841), suggesting their high potential for bioactivity. Among them, we focused on two particularly intriguing compounds, ellagic acid dihydrate and strictinin, which had less than 30 references each, potentially indicating a lack of previous exploration regarding their antioxidant properties (Fig. 5). Ellagic acid dihydrate is a crystalline form of ellagic acid, a well-known polyphenol found in various fruits and nuts, containing two water molecules within its structure. While ellagic acid itself exhibits potent antioxidant activity, exceeding established antioxidants like butylated hydroxytoluene and vitamin E in inhibiting lipid peroxidation [25], the specific activity of its dihydrate form remains unexplored. Given the established bioactivity of ellagic acid, its dihydrate form is a promising candidate for further investigation with a high probability of exhibiting antioxidant properties. Strictinin, a hydrolyzable ellagitannin, has also been demonstrated to possess significant antioxidant properties [26]. Multiple studies have demonstrated that strictinin possesses potent antioxidant properties that can inhibit lipid peroxidation and scavenge free radicals [27,28]. Collectively, our ML approach effectively prioritizes promising candidates for further investigation of their potential antioxidant bioactivities.
Fig. 5
Distribution of publication counts and number of structurally similar active compounds for top-scoring compounds.
Blue bars represent the number of references associated with the top 20 compounds identified through RF and SVM models. The red bars represent the number of structurally similar active compounds present within the training datasets for these same top-ranked compounds. The compounds were ranked based on their average scores obtained from both models. RF, Random Forest; SVM, Support Vector Machine.
![]()
DISCUSSION
This study evaluated the applicability of various ML algorithms for predicting the antioxidant activity of compounds solely based on their chemical structure information. In the current study, we aimed to investigate a baseline performance for these models within a standardized framework. To facilitate a controlled initial assessment, all models were evaluated using their default hyperparameter settings. Under these conditions, RF and SVM demonstrated superior performance compared to other algorithms LR, XGB, and DNN. Notably, DNN displayed the lowest performance among the five models. This finding aligns with the known susceptibility of DNNs to overfitting on datasets with limited sample sizes. The relatively small size of our dataset likely contributed to its underperformance, emphasizing the critical role of data availability and characteristics in model selection. For robust model comparisons, future research should incorporate a rigorous hyperparameter tuning process to optimize the potential of each algorithm.
To simply focus on the applicability of ML in predicting antioxidant activity based on chemical structure, we utilized structural features, particularly ECFP-4 fingerprints (a widely employed representation of compound structure). These fingerprints effectively captured subtle yet critical structural features associated with antioxidant activity. Future research should incorporate feature importance analysis to identify and interpret the most significant features influencing the models' predictions in the context of antioxidant activity. Expanding the feature space to include additional data sources, such as chemical descriptors and chemical-induced transcriptomic data, alongside ECFP-4 fingerprints, could be explored to enhance model generalizability and provide a more comprehensive understanding of the structure-function relationship in antioxidant activity.
While in silico approaches offer significant promise, experimental validation remains an essential step. Compounds predicted by the ML models to have high antioxidant activity should be subjected to biochemical assays to confirm their activity. This step is critical for bridging the gap between computational predictions and practical applications, ensuring that predictions translate reliably into real-world benefits. We propose that our models can serve as a preliminary screening tool, facilitating the selection of candidate compounds for subsequent in vitro validation.
SUPPLEMENTARY MATERIALS
Supplementary data including one figure and one table can be found with this article online at https://doi.org/10.4196/kjpp.2024.28.6.527
 XML Download
XML Download