

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Cardiovascular disease is a leading cause of mortality worldwide [1]. Mainly, cardiovascular disease caused by atherosclerosis, a chonic inflammatory disease of vessel [2]. Numerous studies indicated that toxic agents come from diets, smoke, or oxidized low-density lipoprotein was induced inflammation in various organs [3-5]. Structurally, after various cells are increased levels of pro-inflammatory cytokines or chemokines releasement, its-immune response accelerates immune cell developments [6]. Macrophages are one of the major cell types of the immune systems and regulates inflammation and infection though antigen presentation, polarization, and phagocytosis [1]. Activated macrophages are secreted cytokines or nitric oxides that induces endothelial cell (EC) dysfunction and increased number of abnormal vascular smooth muscle cell [7-9]. In that times, macrophage-derived matrix metalloproteinase-9 (MMP9) expression was involved in macrophage infiltration by destroying collagen in EC [10]. Finally, complex of MMPs and macrophage activation increased development of atherosclerotic plaque formation [10].
Lipopolysaccharides (LPS) has been used for inducing inflammation-mediated damage models in liver, kidney, lung, and EC [4,5,8,11,12]. Mechanistically, LPS lead to activation of inflammatory signaling pathway including mitogen activated protein kinase (MAPK), protein kinase B (Akt), and nuclear factor kappa-light chain enhancer of activated B cells (NF-kB) [8]. p38/MAPK, one of the MAPKs, plays a significant role in inflammation by releasing inflammatory mediators such as tumor necrosis factor-a interleukins (ILs), and monocyte chemoattractant protein-1 (MCP-1) [11]. Activation of p38 also contributes to macrophage polarization and migration though Akt phosphorylation, exacerbating the progression of atherosclerosis [11].
Reactive oxygen species (ROS) can lead to immune dysfunction and impaired cell proliferation when deficient in the cell. However, the excessive release of superoxide radicals, generated by macrophages activated due to cellular oxidative stress, can lead to apoptosis, aging, and inflammatory diseases [7]. Therefore, antioxidants are used to protect cells and tissues from high concentrations of ROS caused by oxidative stress. Nuclear factor erythoid 2-related factor 2 (Nrf2), a transcriptional regulator of intracellular oxidative stress, undergoes increased translocation into the nucleus in response to inflammation-stimulating factors. This translocation leads to the release of antioxidants, which can inhibit ROS and suppress inflammation [13].
Sialyllactose (SL) is the one of the components on human milk oligosaccharides, which is the third major solid component in breast milk, are recognized as the first beneficial nutrient in infants [14]. There are two different structures of SLs that 3'-SL and 6'-SL which is recognized by linking with monosaccharides N-acetylneuraminic acid on and the galactosyl subunit of lactose at the 3' or 6' position, respectively [11]. Our group have been reported that 3'-SL or 6'-SL exert a wide range of pharmacological effects on acute liver injury, EC hyperpermeability, and phenotypic switching of vascular smooth muscle cell [3,4,8,15]. However, the effects of 6'-SL on LPS-induced macrophage activation on atherosclerosis progression and its-related signaling pathway remain unclear. This study aimed to investigate the effect of 6'-SL on LPS-induced macrophage activation in in vitro and in vivo.
METHODS
Reagents and antibodies
Rabbit antibodies against phosphorylation NF-kB p65 (Ser546) (#3033S), NF-kB (#8242), phosphorylation Akt (Ser473) (#4058), Akt (#2685), phosphorylation p38 (Th180/Th182) (#4631), p38 (#8690), Nrf2 (#12721), poly ADP-ribose polymerase (PARP) (#9532) were purchased from Cell Signaling Technology, Inc. Rabbit antibodies against MCP-1 (sc-28879) and mouse antibodies against b-tubulin (sc-166729) were purchased from Santa Cruz Biotechnology. Rabbit antibody MMP9 (AB19016) was purchased from Merck Milipore. Rat antibody against vascular endothelial-cadherin (VE-cadherin, #555289) was purchased from BD Biosciences. 6'-SL salt powder was purchased from GeneChem Inc. LPS (from Escherichia coli O111:B4, EL2630) was purchased from Sigma-Aldrich. SB203580 (S7741), an inhibitor of p38, and LY294002 (S1105), and inhibitor of Akt were purchased from Selleckchem. Phosphate buffered saline (PBS, #EBA-1105) was purchased from Elpisbio.
Cell culture
The murine macrophage cell line, RAW 264.7 (#40071), was procured from the Korean Cell Line Bank. Cells were maintained in Dulbecco’s Modified Eagle’s Medium (Gibco), supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin (100 U/ml, Gibco), and incubated at 37°C in a 5% CO2 humidified environment.
Cell viability
RAW 264.7 cells were plated in a 96-well plate and then incubated at 37°C with 5% CO2 for 24 h. Subsequently, the cells were pre-treated with varying concentrations of 6'-SL for 1 h before being stimulated with 1 μg/ml of LPS for another 24 h. Cell viability was assessed using the 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyltetrazolium bromide assay (MTT, Sigma-Aldrich), with absorbance readings taken at 570 nm using a microplate reader (SpectraMax iD3; Molecular Devices).
Measurement of ROS level
ROS production was assessed using the dihydroethidium (DHE) assay (#D11347, Invitrogen). RAW 264.7 cells were seeded in 12-well plates and incubated overnight at 37°C with 5% CO2. Following treatment, the cells were incubated with 10 μM DHE in PBS for 45 min at 37°C. Subsequently, the cells were washed, fixed in 4% formaldehyde for 10 min, and then washed again and kept in 1X PBS. Fluorescence images of DHE-stained cells were captured using a fluorescence microscope (Olympus IX 71; Olympus). The intensity of DHE fluorescence was analyzed using ImageJ software.
Nuclear and cytoplasmic extraction
RAW 264.7 cells were collected using PBS and centrifuged at 13,000 rpm for 1 min at 4°C. Nuclear extraction was conducted following a previously reported method with minor adjustments [16]. Cells were incubated with a hypotonic buffer containing 100 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (pH 7.9), 1 M KCl, 100 mM 1,4-Dithiothreitol (DTT), and 1X protease inhibitor cocktails for 5 min. Subsequently, 10% NP-40 was added, and the mixture was incubated on ice for 15 min. The samples were then centrifuged at 5,000 rpm for 5 min, and the resulting supernatant contained cytoplasmic proteins. The pellet was washed with PBS, resuspended in a hypertonic buffer comprising 100 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (pH 7.9), 2.5 M NaCl, 100 mM DTT, and 1X protease inhibitor cocktails, and incubated on ice for 15 min. After centrifugation at 13,000 rpm for 10 min, the supernatant was collected as nuclear protein. Finally, 20 μl of the samples were loaded onto sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gels.
Western blot (WB) analysis
The cells were lysed with a 2X SDS buffer consisting of 100 mM Tris–HCl (pH 6.8), 4% SDS, 20% glycerol, 200 mM β-mercaptoethanol, and 0.02% bromophenol blue. The protein extracts were then separated by SDS-PAGE and analyzed by western blotting, following previously established protocols [17].
Quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR)
The qRT-PCR assay was performed according to previous report by minor modification [18]. After treatment, total RNA was isolated using a Tri-RNA reagent (Favorgen) and subsequently synthesized to complementary DNA by the reverse transcription 5X master mix (Eplisbio). The primer IL-1b, MCP-1, and heme oxygenase-1 (HO-1) were synthesized by BIONNER. Primer sequences are listed in Table 1. The relative gene expressions were calculated using the 2–∆∆ct method, normalized by the housekeeping gene b-actin.
Luciferase reporter assay
Cells were co-transfected with pNF-ĸB-Luc or antioxidant response element (ARE)-Luc promoter and Renilla-Luc plasmid by the Solfect (Biosolyx) according to our previous report [5]. After transfection, cells were pretreated with 100 or 200 μM 6'-SL, 10 μM LY294002, or 10 μM SB 203580 for 1 h, followed by treatment with 1 mg/ml LPS for 12 h. Luciferase activity was evaluated by a dual-luciferase reporter assay system (E1960; Promega BioSystems Sunnyvale Inc.).
Immunofluorescence (IF) analysis
IF analysis was performed by the following previous report with minor modifications [19]. RAW 264.7 cells were seeded onto glass coverslips and treated with 1 μg/ml LPS, 100 or 200 μM 6'-SL, 10 μM LY294002, or 10 μM SB 203580 for 24 h in a 12-well plate. Cells were rinsed with PBS twice and fixed 4% paraformaldehyde for 10 min. Then, 0.2% Triton X-100 (JUNSEI) was added for 10 min for permeabilization at room temperature (RT). After blocking, RAW 264.7 cells were incubated with primary antibody against Nrf2 (1:200 dilution) overnight at 4°C and incubated with Alexa Fluor 488-conjugated secondary antibodies (1:250 dilution) for 1 h at RT. Finally, all cells were mounted with 4',6-diamidino-2-phenylindole (DAPI) solution, and images were obtained using a K1-Fluo laser scanning confocal microscope (Nanoscope Systems). Fluorescence values were detected and merged using ImageJ.
Animals
Male ICR mice were obtained from SAMTAKO Inc. Mice were allowed to acclimate to the environment for one week before the start of the experiments and fed standard mice feed at 23°C ± 2°C, humidity (50% ± 5%), under a 12:12 dark:light cycle. All animal experiments were approved by with the Institutional Animal Care and Use Committee of Chungnam National University (202304A-CNU-063). Mice were randomly divided into 4 groups: group1, control; group 2, 10 mg/kg LPS; group 3, 100 mg/kg 6'-SL; group 4, 100 mg/kg 6'-SL + 10 mg/kg LPS. Mice were intraperitoneally (i.p.) administered 100 mg/kg 6'-SL for 2 h, except those in group 1 and 2. After 2 h, the mice were injected with 10 mg/kg LPS diluted in PBS via and i.p. injection for 6 h. Finally, the mice were sacrificed and aorta was collected.
En face IF staining
En face staining was employed to determine the expression of MCP-1 and MMP9 in mouse aortic endothelium according to a previously described method [12]. Mouse aortas were perfused with pre-chilled PBS followed by 4% paraformaldehyde. The harvested aortas were cleaned of fat and connective tissues, cut longitudinally, permeabilized with 0.2% Triton X-100 for 10 min, and blocked with 3% bovine serum albumin for 30 min at RT. Following the blocking process, cross-reactivity of primary antibodies with secondary antibodies was performed, in particular, aortas were incubated with primary antibodies against MCP-1 or MMP9 with VE-cadherin (1:250 dilution) overnight at 4°C and incubated with Alexa Fluor 488- or 546-conjugated secondary antibodies (1:250 dilution) for 1 h at RT. Finally, all aortas were mounted with DAPI solution, and images were obtained using a K1-Fluo laser scanning confocal microscope (Nanoscope Systems).
ROS determination on aortic endothelium
We performed en face staining to evaluate ROS expression in the mouse aortic endothelium according to our previous paper [8]. After sacrifice, mice were perfused with PBS to drain blood and harvest the thoracic aorta. The harvested aorta was cut open longitudinally, incubated with 40 mM DHE reagent in PBS for 60 min at 37°C, and washed with PBS. The aortas were then fixed with 4% paraformaldehyde for 10 min, washed with PBS, fat removed, and permeabilized with 0.2% Triton X-100 for 10 min. After washing with PBS, aortas were stained with DAPI (indicated nucleus). Images were acquired using a K1-Fluo laser scanning confocal microscope (Nanoscope Systems).
Statistical analysis
Statistical analyses were performed using GraphPad Prism 5 (version 5.02; GraphPad Software Inc.). One-way analysis of variance followed by Bonferroni multiple comparisons or two-tailed student's t-test was used for two group comparisons were performed. performed. All data are expressed as the mean ± standard error of the mean and all experiments were performed independently at least 3 times. p < 0.05 was considered statistically significant.
RESULTS
Effect of 6'-SL on cell viability and Akt and p38 signaling pathway in RAW 264.7 cells
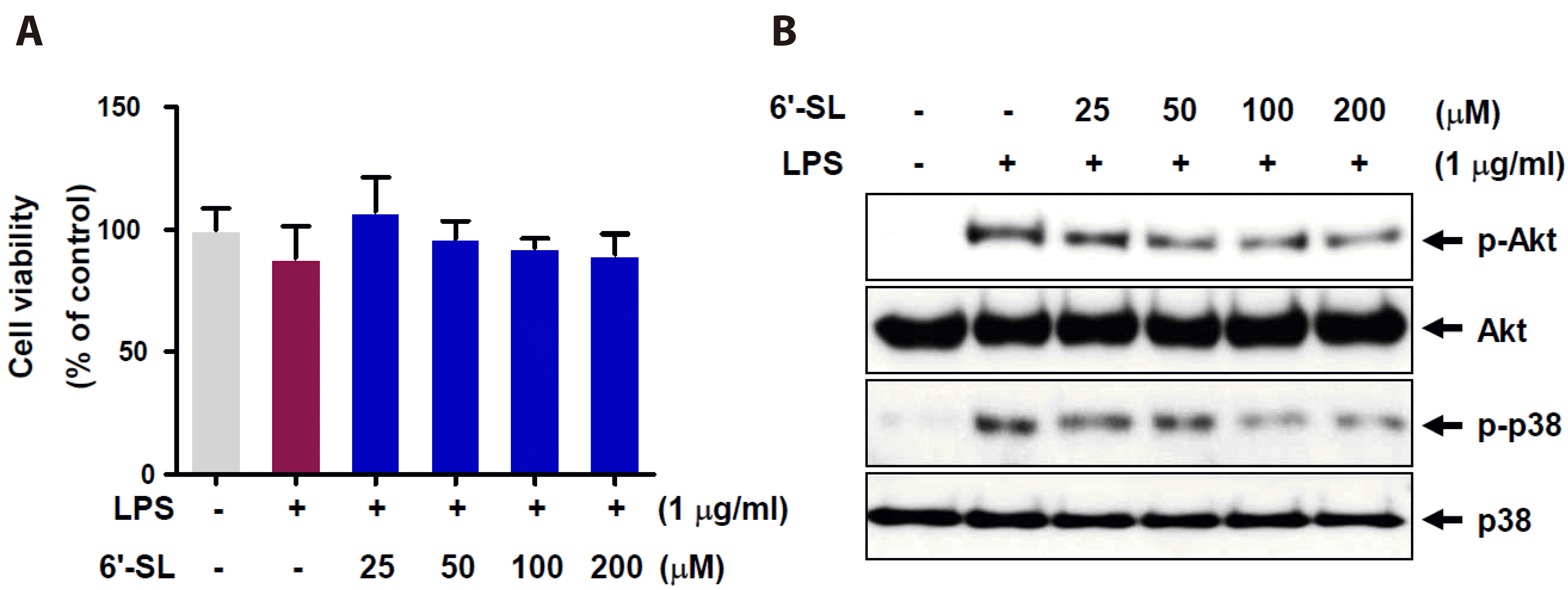
To investigate the effect of 6'-SL on cell survival in LPS-stimulated RAW 264.7 cells, cells were pretreated with 25, 50, 100, and 200 mM of 6'-SL for 1 h, followed by treatment with 1 mg/ml of LPS for 24 h after that cell viability was measured by MTT assay. As shown in Fig. 1A, both LPS stimulation and 6'-SL treatment demonstrated no cytotoxicity on RAW 264.7 cells. Next, we assessed the phosphorylation of Akt and p38 to evaluate inflammatory signaling pathways in the cells. LPS treatment led to increased phosphorylation levels of Akt and p38, while 6'-SL decreased the phosphorylation levels of both proteins in a dose-dependent manner compared to the LPS-treated group (Fig. 1B).
6'-SL inhibits LPS-induced inflammation by suppressing NF-κB translocation and transactivation in RAW 264.7 cells
The nuclear expression of NF-κB plays a pivotal role in the function of inflammatory mediators as a transcription factor [15]. To investigate whether 6'-SL inhibits the phosphorylation and translocation of NF-κB, cells were pretreated with 25, 50, 100, and 200 μM of 6'-SL for 1 h, followed by treatment with 1 μg/ml of LPS for 24 h. The activation of NF-κB p65 induced by LPS was inhibited by 50 μM of 6'-SL, with its activation steadily suppressed up to 200 μM of 6'-SL (Fig. 2A). The protein expression of PARP was used as a marker for nuclear extraction (Fig. 2B). Nuclear expression of NF-κB was completely abolished by pretreatment with 25 μM of 6'-SL (Fig. 2B), and this inhibitory effect was further enhanced by 200 μM of 6'-SL pretreatment (Fig. 2B). Consistent with the nuclear expression of NF-κB, the promoter activation of NF-κB was significantly increased by LPS, approximately 2-fold compared to the control (Fig. 2C). Interestingly, pretreatment with 200 μM of 6'-SL significantly suppressed LPS-induced NF-κB promoter activation to a similar extent as LY294002 (Fig. 2C). MMP9 is an enzyme crucial for macrophage migration, and its effects contribute to increased immune cell infiltration in the progression of atherosclerosis [2]. As shown in Fig. 2D, LPS stimulation markedly enhanced MMP9 expression, whereas its expression was consistently inhibited by 6'-SL. As downstream molecules of NF-κB and MMP9 in the inflammatory response, mRNA expression levels of IL-1β and MCP-1 were assessed via qRT-PCR. As anticipated, LPS treatment significantly upregulated the mRNA expression of IL-1β and MCP-1 by at least 1.5-fold compared to the control sample (Fig. 2E, F). Interestingly, pretreatment with 100 μM of 6'-SL completely suppressed LPS-mediated IL-1β and MCP-1 mRNA expression compared to the non-treated condition (Fig. 2E, F). Furthermore, this inhibition was comparable to the deactivation of Akt by LY294002 and p38 by SB203580 (Fig. 2E, F).
Antioxidative effects of 6'-SL in RAW 264.7 cells
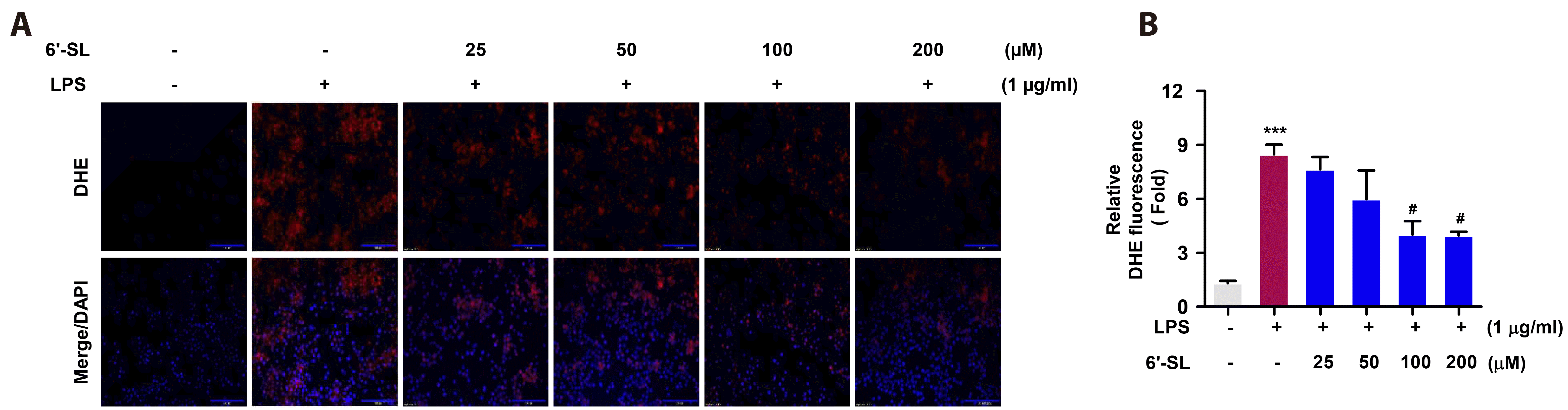
Uncontrolled ROS production regulates multiple cellular mechanisms, including inflammation and cell apoptosis [7]. Therefore, to investigate the role of 6'-SL in LPS-induced ROS production, the treated cells were stained with DHE. LPS stimulation resulted in a 9-fold increase in the fluorescence intensity of DHE compared to the control (Fig. 3A). Treatment with 100 μM 6'-SL significantly reduced LPS-mediated ROS production by approximately 3-fold compared to the LPS-treated condition (Fig. 3B). Nrf2 serves as a key molecule in the antioxidant signaling pathway [14]. To verify this, we treated cells with LPS, SL, and kinase inhibitors, and examined Nrf2 nuclear translocation though IF analysis. As shown in Fig. 4A, Nrf2 clearly translocates into the nucleus in response to all stimuli except LPS. In contrast, LPS stimulation results in a slight increase in cytoplasmic expression of Nrf2 compared to the control (Fig. 4A, B). When Nrf2 expression was analysis by WB, LPS stimulation resulted in increased protein expression of Nrf2 (Fig. 4C). However, pretreatment with 100 and 200 μM of 6'-SL dose-dependently elevated Nrf2 expression levels (Fig. 4C). In the antioxidant signaling pathway, Nrf2 enhances the expression of antioxidant-related genes by binding to the ARE in the promoter region [13]. To investigate how 6'-SL activates the antioxidant signaling pathway, Nrf2 transcriptional activity was evaluated by ARE-luc reporter assay (Fig. 4D) and transcriptional target gene, HO-1 mRNA expression (Fig. 4E). As depicted in Fig. 4D, LPS treatment led to a slight increase in ARE promoter activity, while treatment with 100 μM of 6'-SL significantly enhanced ARE promoter activation. Consistent with the expression of Nrf2, 200 μM of 6'-SL further accelerated the antioxidant signaling pathway by inducing ARE promoter activity (Fig. 4D). In addition, the mRNA expression of HO-1, an antioxidant enzyme produced though Nrf2 transcription, was significantly increased at 200 μM of 6'-SL compared to the LPS-treated group (Fig. 4E).
6'-SL suppresses ROS production in mouse endothelium
To examine the effect of 6'-SL on macrophage-derived ROS production in LPS-activated mouse endothelium, isolated mouse aortas were stained with 40 mM DHE for 1 h, and the fluorescence intensity was measured by confocal microscopy (Fig. 5A). As shown in Fig. 5B, C, administration of LPS for 6 h significantly increased the number of DHE-stained ECs in the mouse endothelium. Consistent to in vitro data, 6'-SL significantly suppressed LPS-induced ROS production, similar to non-injected samples.
6'-SL suppresses ROS production in mouse endothelium 6'-SL inhibits LPS-induced MMP9 and MCP-1 expression in mouse endothelium
To further investigate the role of 6'-SL in LPS-induced macrophage inflammation, mouse aortas were isolated from wild-type ICR mice after i.p. injection with 10 mg/kg LPS for 6 h, and then the expression of MMP9 and MCP-1 was analyzed using en face IF assay. As depicted in Fig. 6A, LPS stimulation significantly increased the expression of MMP9 in EC cytoplasm in the s-flow area. Additionally, injection of 6'-SL significantly suppressed LPS-induced MMP9 expression (Fig. 6A). Consistent with the s-flow data, 6'-SL injection suppressed the LPS-accumulated expression of MMP9 in the d-flow area (Fig. 6B, C). To validate the anti-inflammatory effects of 6’-SL on mouse endothelium, MCP-1 expressions were assessed in both the s-flow and d-flow areas (Fig. 6D–F). LPS treatment increased the number of MCP-1-stained ECs and their expression. Whereas, injection of 6'-SL further inhibited LPS-induced MCP-1 expression in the s-flow area (Fig. 6D–F). Similar to the expression in the s-flow area, 6'-SL suppressed the LPS-induced fluorescence intensity and number of MMP9 and MCP-1-stained ECs in the d-flow area (Fig. 6D–F).
DISCUSSION
Regulating immune cell activation, particularly monocyte-to-macrophage differentiation, plays a pivotal role in inflammation. This is crucial because differentiated macrophages can infiltrate various organs, including the liver, kidney, or vessels, and subsequently release pro-inflammatory cytokines and chemokines [1,11,20]. Mechanistically, during macrophage migration to other cells, the expression of MMPs leads to destabilization of membrane junction protein expression [21]. In atherosclerosis models, overexpression of MMP9 in macrophages enhances acute plaque disruption, thereby increasing macrophage proteolytic activity and promoting plaque instability [21,22].
LPS is known to activate macrophage polarization to M1 or M2 and induce the release of pro-inflammatory cytokines such as tumor necrosis factor-α and IL-1β, as well as ROS or nitric oxide production [23,24]. Previous studies from our group have demonstrated that LPS stimulation leads to mRNA expression of pro-inflammatory cytokines, including IL-1β and MCP-1, though the activation of MAPK, signal transducer and activator of transcription, and NF-κB signaling pathways [4,8,11,15]. When LPS induces an inflammatory response, various signaling pathways are activated, leading to its mediated inflammatory response by enhancing its transcriptional activity [25].
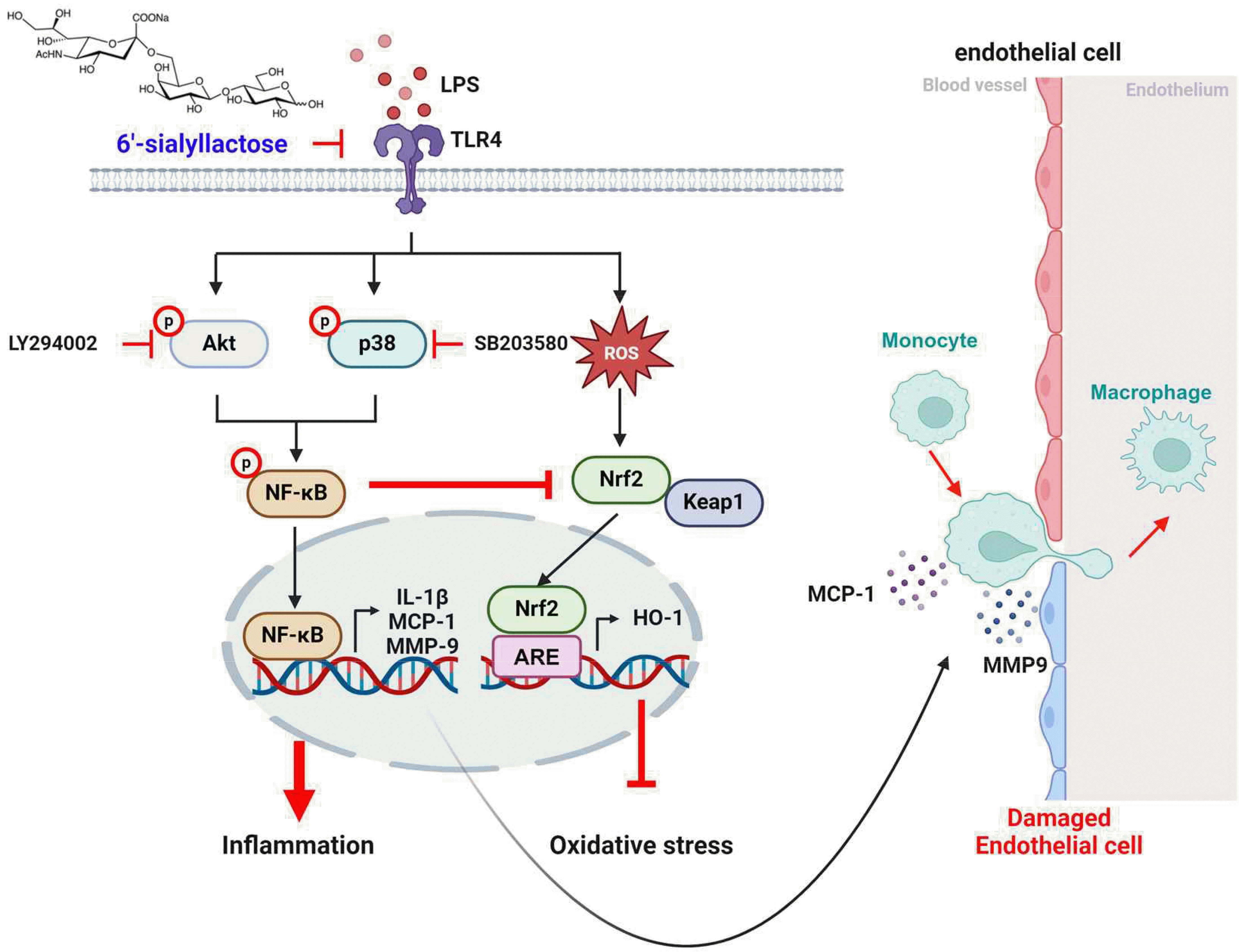
Our data demonstrated that treatment with LPS significantly activates Akt and p38 signaling pathways, whereas, pretreatment with 6'-SL suppresses the phosphorylation of Akt and p38 induced by LPS in a dose-dependent manner, suggesting that 6'-SL has inhibitory effects on these pro-inflammatory pathways (Figs. 1B and 7). As downstream events, we further found that 6'-SL in modulating the NF-κB signaling pathway, a key activator of inflammatory mediators. 6'-SL abolishes the LPS-mediated nuclear expression and promoter activity of NF-κB at 100 μM (Fig. 2A–C).
Moreover, 6'-SL demonstrates dose-dependent inhibition of LPS-induced protein expression of MMP9, a crucial enzyme implicated in inflammatory processes (Fig. 2D). Consistent with this, pretreatment with 100 μM of 6'-SL significantly suppresses the mRNA expression of IL-1β and MCP-1, downstream molecules of NF-κB and MMP9, to a similar extent as inhibitors of Akt and p38 (Fig. 2E, F). In line with the in vitro findings, administration of LPS to mice results in a significant increase in the fluorescence intensity of MMP9 and MCP-1 in both d-flow and s-flow areas of isolated aortas (Fig. 6). However, injection of 6'-SL effectively suppresses the LPS-induced expression of MMP9 and MCP-1 in these areas (Fig. 6).
The balance between oxidant and antioxidant levels is crucial for macrophage polarization [26]. M1-type activation leads to the production of ROS and pro-inflammatory cytokines, which in turn increase the numbers and activation of macrophages [27]. Under non-stress conditions, ROS levels are kept at very low levels and tightly regulated within cells. When ROS levels are increasing by the stimuli, oxidative stress is prevented by the nuclear translocation of Nrf2, an antioxidant [13]. This occurs when Nrf2 dissociates from kelch-like ECH-associated protein 1 (KEAP1), reducing ROS production and repairing ROS-induced damage [13]. However, sustained excessive ROS levels lead to an increase in Nrf2 and KEAP1 in the cytosol but a decrease in Nrf2 nuclear translocation [28]. To verify this, we treated cells with LPS, SL, and kinase inhibitors, and examined Nrf2 nuclear translocation though IF analysis. As shown in Fig. 4A, Nrf2 clearly translocate into the nucleus in response to all stimuli except LPS. In contrast, LPS stimulation results in a slight increase in cytoplasmic expression of Nrf2 compared to the control (Fig. 4A, B). This suggests that the increase in Nrf2 protein expression induced by LPS, observed in the Western blotting data (Fig. 5C), is due to its expression in the cytoplasm rather than its activation in the nucleus. In the nucleus, Nrf2 binds to ARE in the promoter regions of various genes involved in antioxidant defense, detoxification, and cellular protection [13]. This binding induces the expression of these genes, thereby enhancing the cell's capacity to neutralize ROS and mitigate oxidative damage [13]. Therefore, Nrf2 transcriptional activity was evaluated by ARE-luc reporter assay (Fig. 4D) and transcriptional target gene, HO-1 mRNA expression (Fig. 4E). Our data showed that LPS treatment led to a slight increase in ARE promoter activity, while treatment with 100 μM of 6'-SL significantly enhanced ARE promoter activation. Consistent with the expression of Nrf2, 200 µM of 6'-SL further accelerated the antioxidant signaling pathway by inducing ARE promoter activity (Fig. 4D). In addition, the mRNA expression of HO-1, an antioxidant enzyme produced though Nrf2 transcription, was significantly increased at 200 µM of 6'-SL compared to the LPS-treated group (Fig. 4E). Therefore, it suggests that 6'-SL would reduce excessive ROS though antioxidant defense by Nrf2 transactivation and subsequent expression of HO-1.
p38 is a central regulator of the inflammatory response to both exogenous and endogenous injury [29] and contributes to macrophage polarisation and migration though Akt phosphorylation [11]. It has been suggested that there is significant cross-talk between NF-κB and Nrf2 pathways [30,31]. For example, oxidative stress can activate NF-κB, which in turn can lead to an inflammatory response. On the other hand, Nrf2 activation can help to reduce oxidative stress and modulate inflammation, providing a balancing act between these two pathways. Our data indicates that activations of p38 and Akt are involved in NF-κB transactivation and regulates transcriptional targets, such as MMP9, IL-1b and MCP-1 (Fig. 2C–F). Consequently, during the inflammatory response, NF-κB is activated via Akt and p38, which acts to inhibit the activation of Nrf2.
Consistent to in vitro data, administration of 10 mg/kg LPS significantly increased the number of DHE-stained EC and the fluorescence intensity of DHE (Fig. 5C). Furthermore, 100 mg/kg 6'-SL significantly inhibited LPS-induced ROS production to a level comparable to the control condition (Fig. 5C). Together, 6'-SL demonstrates potent anti-inflammatory and antioxidant properties by inhibiting key signaling pathways involved in LPS-induced inflammation and oxidative stress.
Toll-like receptor (TLR)4 is the main receptor for LPS, and upon binding of LPS to TLR4, various cellular mechanisms are activated, leading to an increase in the expression of inflammatory mediator genes [11]. Previously, our group demonstrated that LPS treatment significantly increased the mRNA expression and production of pro-inflammatory cytokines by binding to TLR2 or TLR4 in macrophages or ECs [11] and [12]. Furthermore, pretreatment with 6-SL significantly suppressed LPS-induced TLR4 mRNA expression in ECs [14]. Therefore, it is possible that 6-SL blocks the binding of LPS to TLR4 and its inflammatory signaling pathway. In the future study, we will investigate how 6`-SL suppresses the LPS-mediated inflammatory signaling pathway using an LPS/TLR binding assay. By pursuing this future research plan, we can further elucidate the therapeutic potential of 6-SL as an anti-inflammatory agent and potentially develop novel treatment strategies for inflammatory diseases mediated by TLR4 activation.

 XML Download
XML Download