

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Progressive familial intrahepatic cholestasis (PFIC) comprises a heterogeneous group of rare autosomal recessive disorders characterized by intrahepatic cholestasis typically beginning in childhood. This condition rapidly progresses to cirrhosis and liver failure, often necessitating liver transplantation. To date, six types have been identified [1,2]. PFIC type 2 (PFIC2) results from mutations in the ABCB11 gene, which encodes the bile salt export pump (BSEP) protein responsible for bile acid secretion in the canalicular membrane. These mutations cause an accumulation of bile salts in hepatocytes, leading to progressive liver damage [1,3]. PFIC2 typically manifests in the first few months of life with severe jaundice and pruritus, and progression to end-stage liver disease usually occurs within the first 2 years [2,3]. Although hepatocellular carcinoma (HCC) is more commonly associated with PFIC type 4 (PFIC4), there is a significant risk (up to 15%) of developing HCC or cholangiocarcinoma in patients with PFIC2 [4]. We present a case involving a male child with PFIC2-associated HCC who underwent liver transplantation.
CASE REPORT
This study adhered to the principles outlined in the Declaration of Helsinki. Institutional Review Board (IRB) review was not required for this case report as our institution does not mandate IRB approval for case reports. Written informed consent was obtained for the publication of this case report.
A 3-week-old child presented with jaundice and acholic stools. There was no recorded history of consanguinity. Initially, he showed good weight progression until the age of 2 months, followed by a decline. Subsequently, he developed pruritus, epistaxis, clubbing, and hypertrophic gastritis. The condition progressed to chronic liver disease and cirrhosis, accompanied by portal hypertension, which was further complicated by encephalopathy and pulmonary hypertension.
He exhibited elevated levels of total bilirubin (389.1 μmol/L, reference value <22 μmol/L), conjugated bilirubin (201.9 μmol/L, reference value <5 μmol/L), alkaline phosphatase (785 U/L, reference value 150–380 U/L), aspartate aminotransferase (AST; 390 U/L, reference value 5–60 U/L), and alanine aminotransferase (ALT; 426 U/L, reference value 10–25 U/L). Gamma-glutamyltransferase (GGT) levels were within the normal range (36 U/L, reference value 12–58 U/L). Alpha-fetoprotein levels were significantly elevated (2,300 U/mL). Liver ultrasonography revealed hepatomegaly with a heterogeneous texture and hypoechogenic nodules, some of which were ill-defined. The liver biopsy showed signs of chronic cholestasis, featuring rosettes and xanthomatous transformation in Kupffer cells and hepatocytes, along with a ductular reaction, occasional ductopenia, and cirrhosis. Additionally, several multinucleated hepatocytes and a moderate mononuclear inflammatory infiltrate were observed.
The clinical features, laboratory results, ultrasound, and histological findings were consistent with a diagnosis of PFIC2. Genetic analysis revealed that the individual is homozygous for the V444A polymorphism in the ABCB11 gene. He was initially treated with ursodeoxycholic acid; however, the disease progressed to end-stage liver disease by the age of 2 years, accompanied by the development of the aforementioned liver nodules. Four months later, he underwent transplantation with a cadaveric reduced-size AB0-compatible liver, specifically segments II and III.
Pathological Analysis
Upon gross examination, the native liver appeared dark green and firm, featuring small cirrhotic nodules. Two well-demarcated nodules measuring 2.5 cm and 1.5 cm were prominent on the capsule. Additionally, four other nodules, with diameters ranging from 0.4 to 1.3 cm, were also noted.
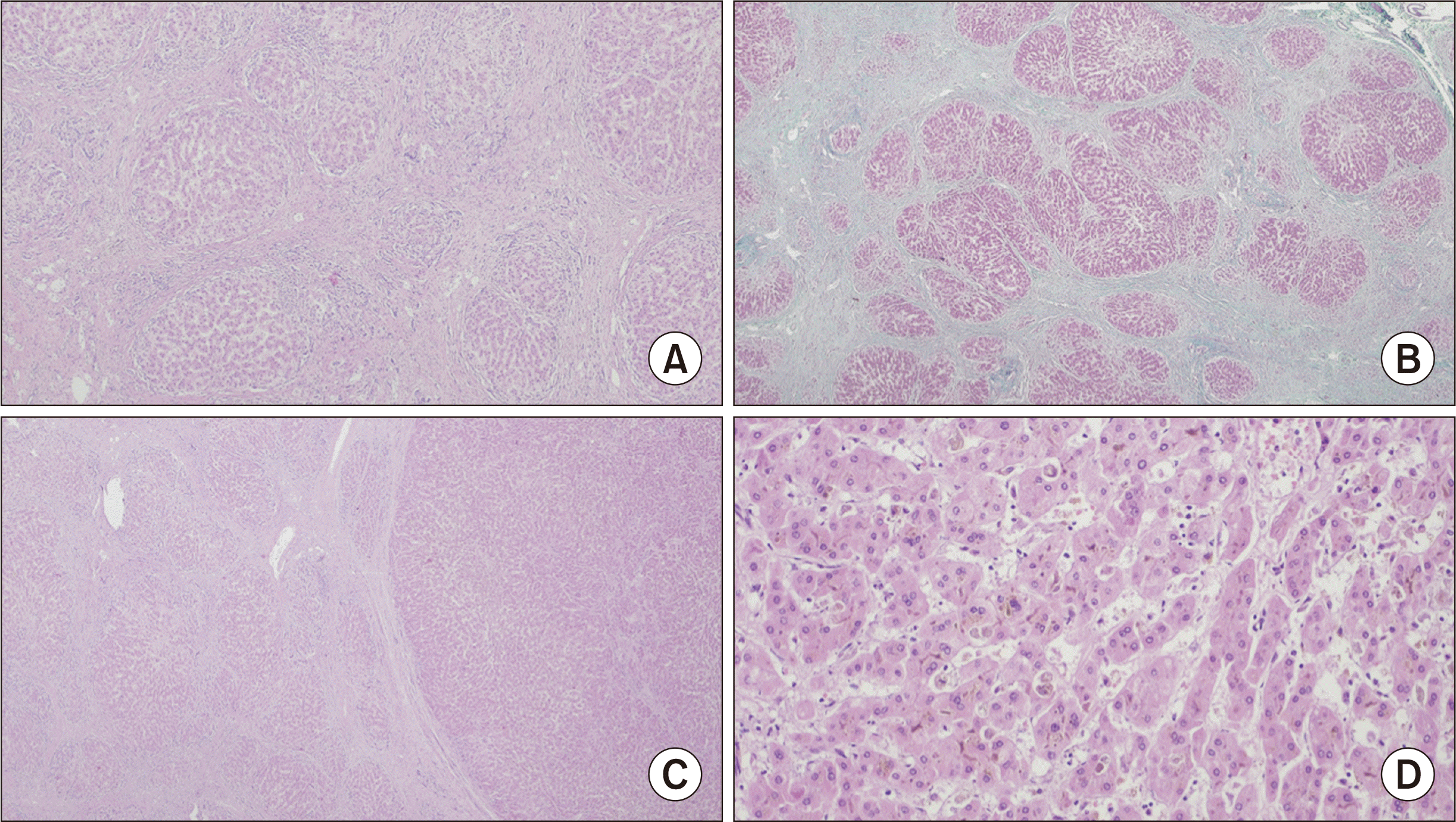
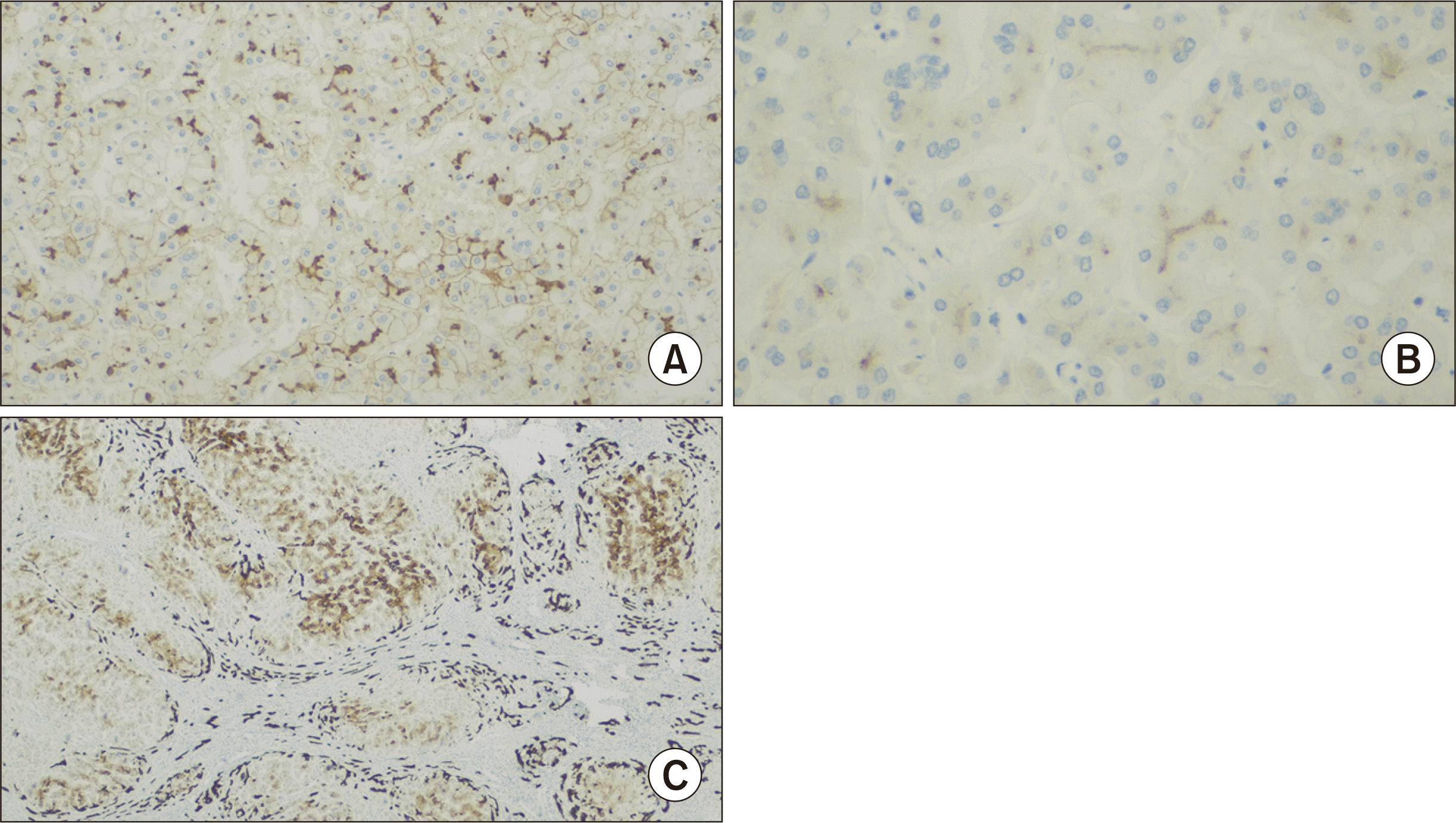
On histological examination (Fig. 1), the liver parenchyma exhibited micronodular cirrhosis along with signs of chronic cholestasis, characterized by numerous Mallory-Denk bodies and copper deposition, as indicated by rhodanine staining. Additionally, a mild septal inflammatory infiltrate, ductular reaction, and ductopenia impacting the interlobular ducts were observed. Immunohistochemistry studies revealed the expression of BSEP, weak expression of multidrug resistance protein 3, and CK7 expression in periseptal hepatocytes (Fig. 2).
The nodules measuring 2.5 cm and 1.5 cm were identified as high-grade dysplastic nodules. The other four nodules were classified as small HCCs with a trabecular pattern, featuring a clear cell component and lacking vascular invasion.
Follow-up
Posttransplant complications included severe cholestasis, persisting until the 16th postoperative day, accompanied by lobular necrosis due to an acute rejection episode (Banff score 3). Additionally, he developed hepatic artery stenosis within the first month following surgery, which required re-anastomosis. He maintains good graft function under immunosuppression with tacrolimus. There have been no episodes of rejection, and no recurrence of disease or neoplasm has been detected, with a follow-up period of 20 years and 9 months.
DISCUSSION
PFIC2 is a rare disease that typically begins in childhood, characterized by an early progression to cirrhosis and liver failure. Additionally, it is associated with HCC and cholangiocarcinoma in approximately 15% of cases [4].
We present a rare case of a PFIC2 patient diagnosed with HCC. The patient exhibited typical symptoms of neonatal jaundice and had normal GGT levels. Rapid progression to end-stage liver disease by the age of 2 is typical in PFIC2 patients [2,3,5]. The biopsy findings, which included cirrhosis, chronic cholestasis, giant cell hepatitis, ductular reaction, and ductopenia, align with those associated with PFIC2 and are consistent with descriptions in the literature [6]. Although immunohistochemistry studies indicated BSEP expression, this could represent a nonfunctional protein. Genetic analysis of the ABCB11 gene revealed only the V444A polymorphism, a variant whose clinical significance remains unclear [7,8].
HCC was detected in the explanted liver following transplantation. This type of neoplasm is uncommon in children, with an incidence rate of approximately 0.41 per million [9]. In patients with PFIC2, only about 15% develop malignancies, predominantly HCC, although there are also reports of cholangiocarcinoma [4]. It is important to note that HCC is not exclusive to PFIC2; cases of HCC associated with PFIC4 have been reported as well [10]. The possibility of developing HCC at a young age should always be considered in PFIC2 patients, particularly when nodules are visible on imaging exams. Instances of HCC have been documented in children as young as 7 months old [11,12].
The primary objectives of treatment are to decelerate the progression of the disease and to alleviate pruritus. Medical strategies encompass the use of ursodeoxycholic acid and other antipruritic medications, including cholestyramine and rifampicin. Surgical interventions, such as biliary diversion techniques, have proven successful in patients with PFIC2. If these approaches prove ineffective, or if complications such as end-stage liver disease or HCC arise, liver transplantation is considered the sole viable option [3,13].
Liver transplantation was curative in this case, with no recurrence of disease or neoplasm observed over a follow-up period exceeding 20 years. There have been reports of cases where HCC led to death, particularly in children who had metastatic disease at the time of diagnosis and were therefore only eligible for palliative treatment [14]. Additionally, a significant number of diagnoses have been made incidentally [14,15]. Given these considerations, early diagnosis is essential. Consequently, routine monitoring for HCC development in PFIC2 patients should be implemented from a very young age, starting possibly within the first year of life, to enable potentially curative interventions in cases of HCC associated with PFIC2.
The development of HCC in children with PFIC2, a neoplasm that is otherwise rare in this age group, should always be considered, and a screening program should be offered. The importance of liver transplantation in treating this disease must be emphasized, as it is currently the only potentially curative treatment available.
 XML Download
XML Download