

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Disorders of GNAS inactivation include pseudohypoparathyroidism types Ia, Ib, and Ic (PHP-Ia, -Ib, and -Ic), pseudopseudohypoparathyroidism (PPHP), progressive osseous heteroplasia (POH), and osteoma cutis (OC). PHP-Ia and PHP-Ic feature hormone resistance and characteristics of Albright hereditary osteodystrophy (AHO), such as short stature and subcutaneous ossification. PHP-Ib is primarily involved in parathyroid hormone resistance. PPHP manifests as AHO without hormone resistance. POH involves progressive bone formation in the muscles and fascia, whereas OC involves ossification of the dermis and subcutaneous tissues. Diagnosis involves identifying the clinical features and genetic or epigenetic alterations affecting the GNAS locus. GNAS inactivation can be caused by pathogenic variants, imprinting issues, epimutations, or paternal 20q disomy. PPHP and POH/OC are caused by pathogenic variants of the paternal GNAS allele [1].
PHP-1b is a rare imprinting disorder characterized by renal parathyroid hormone (PTH) resistance, but the absence of physical features of AHO [2]. This distinguishes PHP-Ib from PHP-Ia, which results from mutations in the GNAS gene encoding the G protein α subunit. The autosomal-dominant form of PHP-Ib (ADPHP-Ib) is linked to chromosome 20q13.3, where the GNAS locus is located. Notably, loss of methylation of exon the A/B differentially methylated region (DMR) of GNAS has been observed in both sporadic PHP-Ib cases and affected members of AD-PHP-Ib families. Recent findings indicate that affected individuals from 12 unrelated AD-PHP-Ib families and four patients with sporadic PHP-Ib, but not healthy controls, are heterozygous for an approximately 3 kb microdeletion approximately 220 kb upstream of GNAS exon A/B. This deleted region includes three exons of the STX16 gene, which does not show any imprinting. Microdeletion leads to loss of methylation at exons A/B, without affecting other GNAS DMRs, suggesting that it disrupts the cis-acting element necessary for exon A/B methylation, causing renal PTH resistance in AD-PHP-Ib [3].
However, there has been no research on the causes of recurrent 3-kb microdeletion. Therefore, we aimed to elucidate the putative mechanism through genomic characterization of the 3-kb microdeletion.
MATERIALS AND METHODS
1. Informed consent and study enrollment
Ten unrelated patients with PHP who visited Seoul National University Hospital between March 2009 and January 2019 were enrolled in the Korean Genetic Diagnosis Program for Rare Diseases Phase II [4]. This study was conducted in accordance with the tenets of the Declaration of Helsinki, and the protocol was approved by the Institutional Review Board of the Seoul National University Hospital. Informed consent was obtained from all enrolled participants.
2. Methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA)
To investigate the genetic and epigenetic changes in the samples, the MS-MLPA technique was employed using the SALSA MSMLPA probemix ME031-B2 GNAS kit, following the manufacturer’ s instructions (MRC-Holland, Amsterdam, Netherlands). Genomic DNA (200 ng) was denatured at 98°C for 5 minutes and hybridized with the ME031-B2 probe mix for 16 hours at 60°C. After hybridization, the product was divided into two tubes for copy number and methylation analyses using a methylation-sensitive endonuclease. The PCR products were analyzed on an ABI 3130xl capillary sequencer (Applied Biosystems, Waltham, MA, USA), and the data were processed using GeneMarker v.1.51 software (SoftGenetics, State College, PA, USA). Peak intensities were normalized to internal control probes, and the intensity ratios of identical probes from the samples were compared with those of the controls.
3. Junction PCR and Sanger sequencing around the breakpoint areas
PCR primers were designed based on the consensus sequences from the split-read junctions. The amplified products were sequenced using an ABI PRISM 3730 xl DNA Analyzer (Applied Biosystems) with the BigDye Terminator v.3.1 Cycle Sequencing Kit (Applied Biosystems). Sequence analysis was conducted using Sequencher software (Gene Codes Corporation, Ann Arbor, MI, USA).
4. Mapping of repetitive elements around the breakpoint areas
Repetitive elements within the breakpoint regions were identified using RepeatMasker in the UCSC Genome Browser (https://genome.ucsc.edu/). Sequence similarities were assessed using ClustalW (https://www.genome.jp/tools-bin/clustalw).
RESULTS
1. Mutation analysis
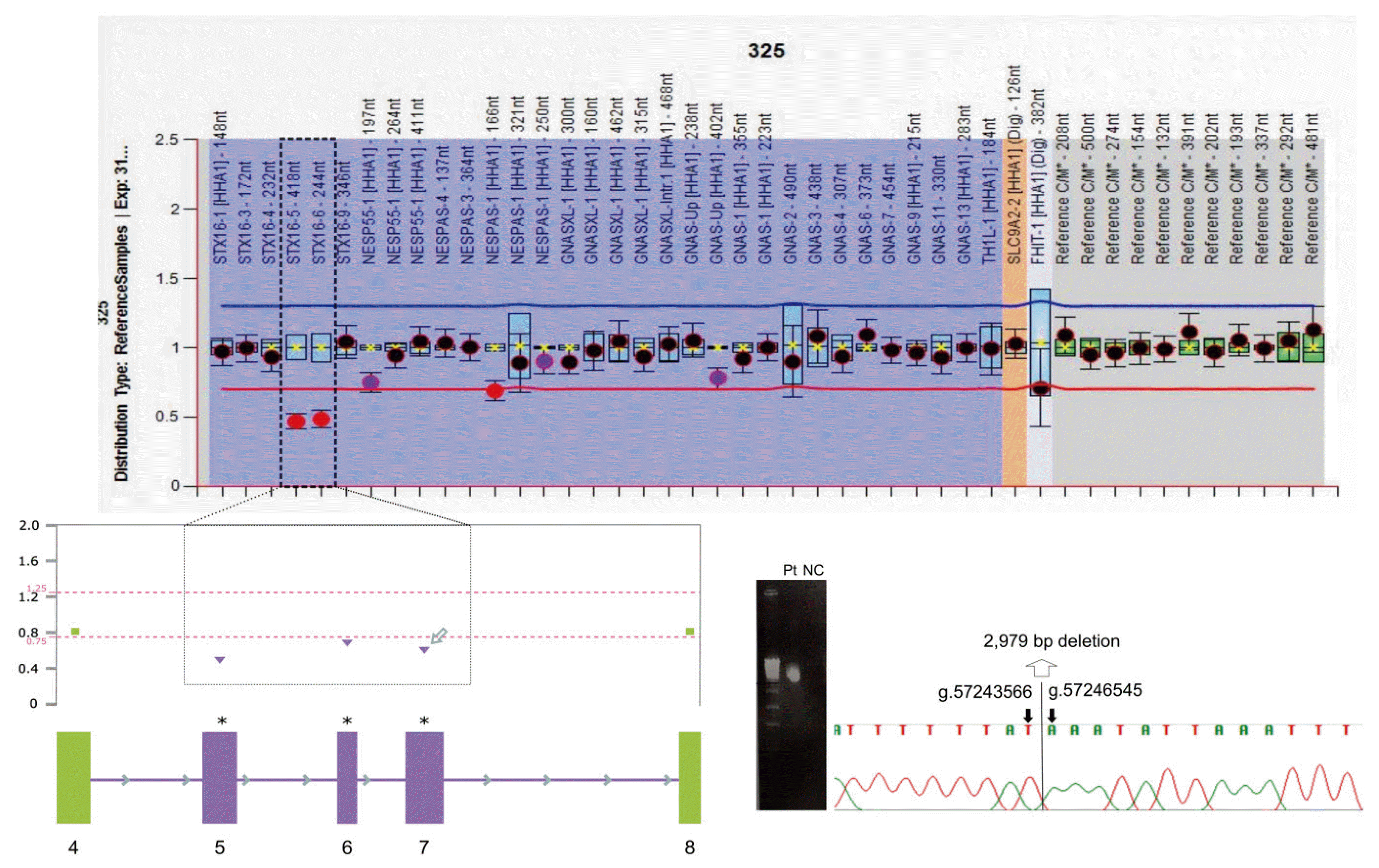
Table 1 summarizes the 10 cases, their classifications, and the genetic testing results. Table 1 comprises data for individuals classified as having PHP-1a and PHP-1b. Genetic testing revealed various abnormalities, such as paternal 20q disomy, STX16 2,979bp deletion, and mutations in the GNAS gene. Some patients exhibited no detectable genetic abnormalities.
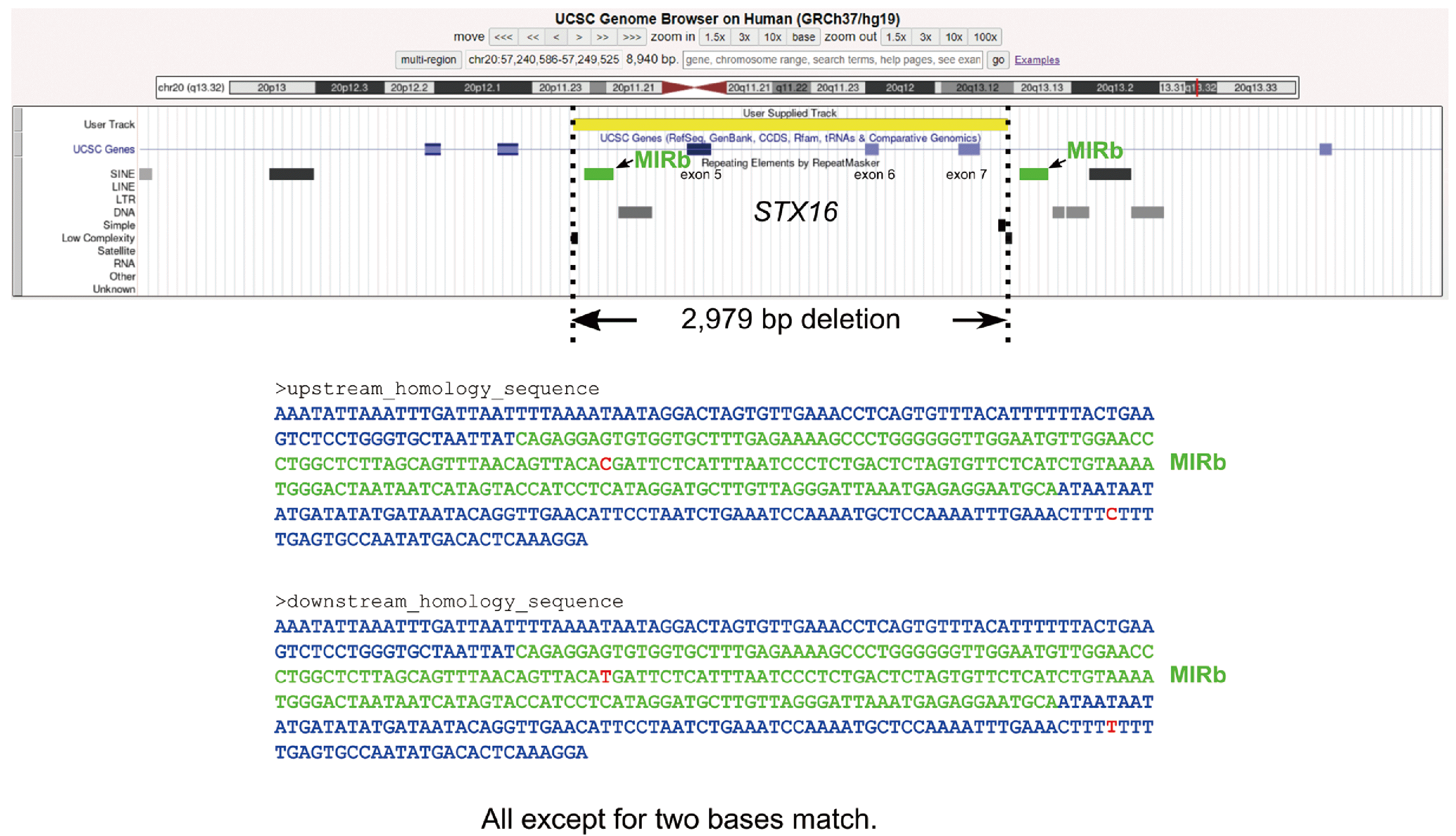
A 2,979 bp deletion of STX16 was detected in two of the 10 probands (20%). Heterozygous deletions in exons 5 and 6 of the STX16 gene were initially identified using MLPA. The deletion of exon 7, which was not detected using MLPA, was suspected based on the results of an in-house copy number variation screening method [5]. Subsequently, junction PCR was performed. Finally, we confirmed a 2,979 bp deletion (chr20:57,243,566–57,246,545) encompassing exons 5–7 (Fig. 1).
2. Junction sequence analyses
When analyzing similarity using ClustalW within 1-kb upstream and downstream of the breakpoints, a 319 bp homologous region was detected, which showed only 2 bp mismatches, as depicted in Fig. 2 (bottom). This region contains one of the mammalian-wide interspersed repeats (MIRs), MIRb, which spans 191 bp. A 1-bp difference was observed between the upstream and downstream MIRb sequences. The upstream homologous sequence was located within intron 4 of STX16, whereas the downstream homologous sequence was located within intron 7.
DISCUSSION
STX16 gene deletion was detected in two of the 10 probands (20%). This deletion range is consistent with previous reports [6-10]. Most genomic rearrangements occur through mechanisms such as non-allelic homologous recombination (NAHR), nonhomologous end joining, fork stalling, and template switching [11]. However, the presence of homologous sequences in proximity to breakpoints favors rearrangements mediated by NAHR [12]. While NAHR mediated by Alu elements is well documented [13, 14], to the best of our knowledge, this is the first cases that NAHR via MIRs is a putative mechanism of rearrangement.
Short interspersed elements are widespread in mammalian genomes. The significant diversity of these repeats across placental orders suggests independent amplification of each lineage following mammalian radiation. Here, we introduce an ancient family of repeats, termed MIRs, whose sequence variation and widespread presence among placental mammals, marsupials, and monotremes indicate amplification during the Mesozoic era. With approximately 120,000 copies still identifiable in the human genome (0.2–0.3% of DNA), MIRs serve as a “fossilized” record of a substantial genetic event preceding the radiation of placental orders [15].
However, the evolutionary role of repeated element sequences in genomes remains debatable. A recent study suggested that an expanded repertoire of repeat elements may enhance organismal fitness by promoting somatic diversity, akin to the beneficial genetic diversity observed in microbial populations. Specifically, the characterization of somatic recombination involving Alu and L1 in this study provides a foundation for future investigations into the dynamics of somatic NAHR events and their implications for genome structure and function [16]. We believe that our study will contribute to broadening our understanding of the mechanisms underlying genomic rearrangements.
In conclusion, the findings of this study suggest that the common 2,979 deletion of STX16 is mediated by MIR-mediated nonallelic homologous recombination.
 XML Download
XML Download