

 PDF
PDF Citation
Citation Print
Print



서 론
체세포 변이 해석의 근거 자료
Table 1
| Database | Website | |
|---|---|---|
| Guidelines | National Comprehensive Cancer Network (NCCN) | https://www.nccn.org |
| World Health Organization (WHO) Classification | https://www.who.int | |
| International Consensus Classification (ICC) | - | |
| European LeukmiaNet (ELN) | https://www.leukemia-net.org | |
| Therapeutic approval | US Food and Drug Administration | https://www.fda.gov |
| Korean Food and Drug Administration | https://www.mfds.go.kr | |
| Cancer-specific variant database | Catalog of Somatic Mutations in Cancer (COSMIC) | https://cancer.sanger.ac.uk/cosmic |
| cBioPortal | https://www.cbioportal.org | |
| Intogen | https://www.intogen.org/search | |
| Database of Curated Mutations (DoCM) | https://docm.info/ | |
| Cancer Hotspots | https://www.cancerhotspots.org/ | |
| Clinical relevance and/or therapeutic implications | OncoKB | https://www.oncokb.org |
| My Cancer Genome | https://www.mycancergenome.org | |
| Clinical Interpretation of Variants in Cancer (CiVIC) | https://civicdb.org/ | |
| Jackson laboratory | https://ckb.jax.org/ | |
| Cancer Genome Interpreter (CGI) | https://www.cancergenomeinterpreter.org/ | |
| MetaKB | https://search.cancervariants.org | |
| ClinicalTrials.gov | https://clinicaltrials.gov |
![]()
1. 인구집단 데이터베이스
2. 종양 관련 데이터베이스
3. 의학 연구 문헌
4. 전산 예측 알고리즘
체세포 변이의 발암성 해석 지침
1. 체세포 변이의 발암성 해석 근거에 관한 점수 산정 및 판정 개요
Table 2
Adapted from Horak et al. [1].
![]()
2. 체세포 변이의 발암성 판정 근거
1) 매우 강한 수준의 근거(Very strong): 8점
i. 매우 강한 발암성(Oncogenic very strong, OVS1): +8점
– 종양억제유전자에서 무효 변이[null variant; 무의미(nonsense), 틀이동(frameshift), 캐노니컬 스플라이스 부위(canonical splice site) ±2 이내, 시작 코돈, 단일 또는 다수의 엑손 결실 변이]가 관찰되는 경우, 매우 강한 수준의 발암성 근거가 된다. 하지만 예상되는 기능 상실 변이(loss of function)가 3’ 말단의 조기 종결 코돈 분해(nonsense mediated decay) 부위에서 발생한 경우에는 그 해석에 주의해야 한다. 또한, 스플라이스 부위 변이가 엑손 결손(exon skipping)을 일으키지만, 틀내 변화(inframe events)와 같이 나머지 단백의 손상이 없는 경우나 종양 억제 기능을 유지하는 잘 알려진 대체 동형단백질(alternative isoform)을 생성하는 경우, 다중 전사(multiple transcripts)에 의하는 경우에는 해석에 주의해야 한다.
2) 강한 수준의 근거(Strong): 4점
i. 강한 발암성(Oncogenic strong, OS): +4점
– OS1: 이전에 잘 확립된 연구에서 발암성으로 알려진 원인 변이와 동일한 아미노산으로의 변이를 일으키는 경우에 부여한다(예, 동일 코돈 내 G>C, G>T로의 변화가 동일한 valine에서 leucine으로의 아미노산 변화를 유발하는 경우). 단, 아미노산 수준의 변화가 아니라 스플라이싱에 영향을 주는 변이인 경우 주의해야 한다.
– OS2: 잘 확립된 체외 또는 체내 기능 연구에서 발암성이 뒷받침되는 경우에 부여한다. 단, 재현 가능하고 견고하게 수립된 연구여야 하며, OS1이 적용되는 경우에는 OS2는 해당 변이와 일치하는 특정 염기서열에 기반한 기능연구에만 적용된다.
– OS3: Cancerhotspots.org에 등재되어 있으며, 최소 50개 이상의 검체에서 동일한 아미노산 위치의 변이가 발생하고, 최소 10개 이상의 검체에서 동일한 아미노산 변이가 관찰된 경우에 부여한다. 단, cancerhospots.org에서 충분히 다뤄지지 않았다면, COSMIC이나 종양 유형에 특화된 연구 자원을 이용할 수 있으며, 특정 조기 종결 돌연변이(truncating somatic variant)에 의한 핫스팟의 경우를 주의해야 한다. OS3는 특정 염기 변화에 근거하여 핫스팟을 관찰할 수 있는 경우가 아니라면, OS1이 적용 가능한 경우에는 사용할 수 없다.
3) 보통 수준의 근거(Moderate): 2점
i. 보통의 발암성(Oncogenic moderate, OM): +2점
– OM1: 변이가 잘 확립된 연구에서 기능적으로 중요한 도메인(효소 활성 부위 등)에 위치한 경우 부여한다. 단, OS1이나 OS3 규칙이 적용 가능한 경우, 이 규칙을 중복하여 적용할 수 없다.
– OM2: 알려진 발암유전자(oncogene)이나 종양 억제 유전자에서 발생한 틀 내 단백길이 변화(in-frame deletions/insertions)를 유발하거나, 종양 억제 유전자에서 나타난 정지 상실 변이(stop-loss variants)의 경우 부여한다. 단, OVS1이 적용가능한 경우 이 근거는 적용할 수 없다.
– OM3: Cancerhotspots.org에 등재되어 있으며, 50개 미만의 검체에서 동일한 아미노산 위치에서의 변이가 발생하고, 최소 10개 이상의 검체에서 동일한 아미노산 변이가 관찰된 경우 부여한다. 단, cancerhospots.org에서 충분히 다뤄지지 않았다면, COSMIC이나 종양 유형에 특화된 연구 자원을 이용할 수 있다. 이 근거는 OM1이나 OM4와 중복하여 적용할 수 없으며, 특정 조기 종결 돌연변이에 의한 핫스팟의 경우를 주의해야 한다.
– OM4: 같은 위치의 아미노산에서의 과오변이가 발암성으로 확인된 경우 부여한다. 단, 아미노산 변화는 발암성으로 알려진 아미노산의 변화보다 더 크거나 최소한 같은 수준의 변화가 일어나야 한다(예, p.Arg156His 변이가 발암성으로 알려졌으나, p.Arg156Cys 변이가 관찰된 경우). 이 근거는 OS1, OS3, 또는 OM1과 중복하여 적용될 수 없으며, 아미노산이나 단백 수준의 변화보다 스플라이싱에 영향을 미치는 경우 주의해야 한다.
4) 약한 수준의 근거(Supporting): 1점
i. 약한 발암성(Oncogenic supporting, OP): +1점
– OP1: 모든 전산 예측(보존/진화 부위, 스플라이싱 효과 등)이 발암성을 뒷받침할 경우 부여한다. 단, 많은 인실리코 알고리즘이 예측을 위해 동일하거나 유사한 입력을 이용하기 때문에 각 알고리즘을 독립적인 기준으로 계산하거나, 여러 번 반복평가되어서는 안된다.
– OP2: 단일 유전 기원을 가지는 종양에서 해당 유전자의 변이가 관찰된 경우 부여한다(예, RB1의 양대립유전자[bi-allelic] 비활성이 유발하는 망막아세포종). 단, 일부 소수의 사례는 다른 메커니즘에 의해 발생할 수 있으며, 조직학적 유사성은 오진을 야기할 수 있다.
– OP3: Cancerhotspots.org에 등재되어 있으며, 10개 미만의 같은 아미노산 변이가 관찰된 경우 부여한다. 단, cancerhospots. org에서 충분히 다뤄지지 않았다면, COSMIC이나 종양 유형에 특화된 연구 자원을 이용할 수 있으며, 특정 조기 종결 돌연변이에 의한 핫스팟의 경우를 주의해야 한다.
– OP4: gnomAD에서 빈도가 관찰되지 않거나, 매우 낮은 빈도로 관찰된 경우 부여한다. 단, 인구집단 데이터베이스에는 삽입/결실 변이가 과소평가되어 있으며, 클론성 조혈 변이들이 포함되어 있을 수도 있음을 고려해야 한다.
3. 혈액암의 주요 체세포 변이
Table 3
Adapted from NCCN guideline V1.2024 MDS and 2022 WHO classification for myeloid neoplasms [2].
![]()
체세포 변이의 임상적 적용가능성 해석 지침
1. 체세포 변이의 임상적 적용가능성의 분류 근거
1) Level A: US FDA 승인된 치료에 대한 반응 및 저항성 또는 특정 암종에 대한 전문적인 가이드라인에 제시되어 있는 치료적, 진단적, 예후적 바이오마커인 경우 해당한다.
2) Level B: 잘 설계된 연구를 기반으로 한 치료에 대한 반응 및 저항성 또는 특정 암종에 대한 잘 설계된 연구에서 제시된 진단적, 예후적 바이오마커인 경우 해당한다.
3) Level C: FDA 또는 다른 전문가집단에서 타암종에 대해 승인된 치료에 대한 반응 및 저항성 또는 다수의 소규모 연구들에 기반한 진단적, 예후적 바이오마커인 경우 해당한다.
4) Level D: 전임상단계의 연구를 기반으로 한 치료적 유용성 또는 소규모 연구나 다수의 증례 보고를 기반으로 한 진단적, 예후적 바이오마커인 경우 해당한다.
2. 체세포 변이의 임상적 적용가능성의 판정 근거
Table 4
![]()
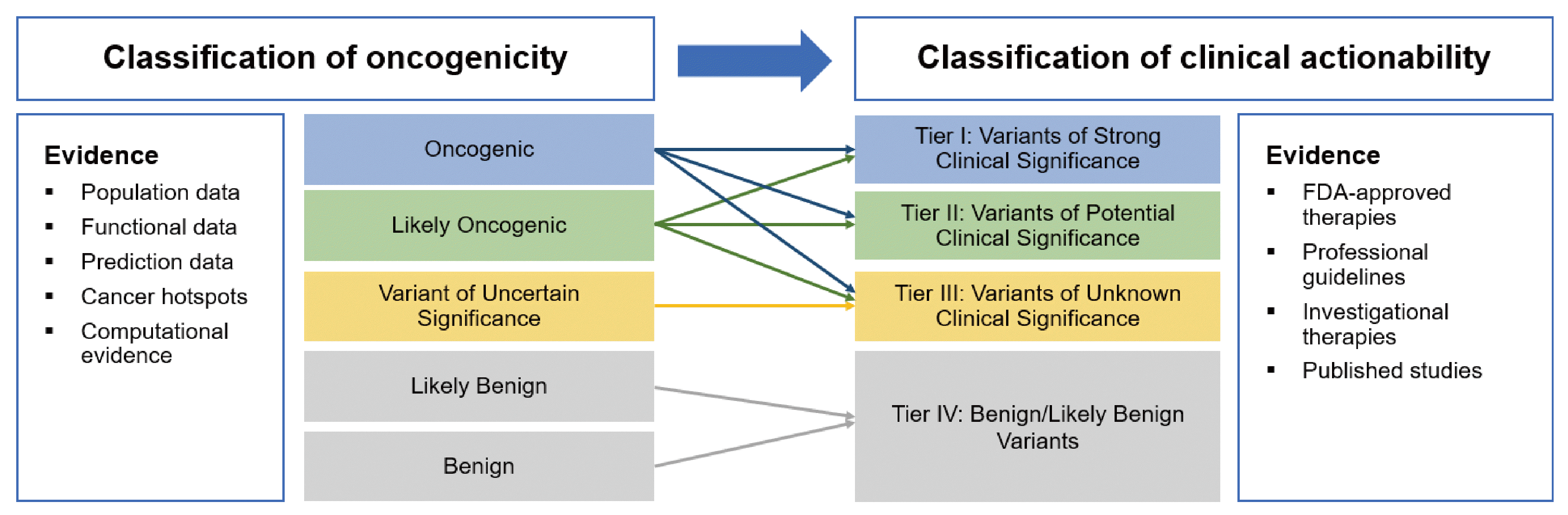
1) Tier I 변이: 강한 임상적 의미를 갖는 변이(variants of strong clinical significance)로 level A와 B의 근거를 갖는 경우에 해당한다.
2) Tier II 변이: 잠재적 임상적 의미를 갖는 변이(variants of potential clinical significance)로 level C 또는 D의 근거를 갖는 경우에 해당한다.
3) Tier III 변이: 임상적 중요성이 알려지지 않은 변이(variants of unknown clinical significance)인 경우에 해당한다.
4) Tier IV 변이: 양성(benign) 또는 준양성 변이(likely benign variants)에 해당한다.
체세포 변이의 분류에 대한 기타 가이드라인
1. 2018 ESMO Scale for clinical actionability of molecular targets (ESCAT)
2. 2019 벨기에 체세포 변이 분류 가이드라인(Belgian somatic variant classification guideline)
3. 2020 스페인 체세포 변이 분류 가이드라인(Spanish somatic variant classification guideline)
혈액암의 체세포 변이 해석 시 고려 사항 및 해석 예시
1. 고형암의 체세포 변이 해석과의 차이점
2. 클론성조혈증(Clonal hematopoiesis of indeterminate potential, CHIP)
3. 여러 변이의 조합에 대한 통합적인 해석
4. 체세포 변이 해석의 예시
1) TP53 NM_000546.6:c.845G>T; p.Arg282Leu
2) ASXL1 NM_015338.6:c.1934dup; p.Gly646Trpfs*12
혈액암의 체세포 변이 보고 지침
1. 체세포 변이의 보고 지침
1) 변이 보고범위
2) 변이의 명명
3) 생식세포 변이의 보고
4) 검출 변이의 임상적 중요성에 대한 기술
5) 검사법 기술
6) 보고서 구성 요소
- 변이의 분류, 유전자 변이, 전사체 참조서열(transcript reference sequence), 변이 대립유전자 빈도(VAF), 커버리지, 검출된 변이에 대한 해석적 기술, 검사방법, 검출한계, 최소 시퀀싱 커버리지, 주요 품질 지표, 시퀀싱 플랫폼, 데이터 분석 파이프라인, 참조 유전체 버전, 분석 유전자/유전자 부위, 변이 분류 방법, 검사의 한계
 XML Download
XML Download