

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease with a broad spectrum of clinical manifestations, ranging from mild joint pain to life-threatening damage to various organs such as kidney, central nervous, and cardiovascular system. SLE is characterized by the production of various autoantibodies to numerous cellular constituents, especially nucleic acids and proteins [1]. SLE develops in genetically susceptible individuals exposed to environmental, sex-related, or endogenous triggers.
Geographic variations contribute to the diversity in the risk and severity of SLE. The SLE incidence rates vary across regions, ranging from 1.5 to 7.4 per 100,000 person-years in Europe, 1.4 to 6.3 in South America, 3.7 to 49.0 in North America, and 2.5 to 8.6 in Asia [2]. Moreover, the prevalence of SLE varies significantly based on ethnicity rather than geographic distribution. Indeed, the prevalence of Asian, Hispanic, and Black African populations is 2-fold higher than that of European population [2,3]. The risk of developing lupus nephritis (LN), one of the leading causes of mortality, was the highest in Asian population [4].
There are still many unmet needs in treating SLE [5,6]. Current treatments for SLE, including corticosteroids, cytotoxic drugs, and immunosuppressants, present various side effects. Thus, refractory disease manifestations, such as nephritis, alongside neurological involvement, require new drugs with greater efficacy and safety. However, most clinical trials over the past 60 years that tested the effectiveness of novel drugs for SLE have yet to be successful. One reason may be the heterogeneity of the disease associated with genetic variants in different races. Several clinical trials on lupus patients showed different treatment responses across races. Sub-analyses in the study of mycophenolate mofetil (MMF) vs. intravenous cyclophosphamide (IVC) as induction therapies of LN revealed that statistically significantly fewer patients responded to IVC compared to MMF in mixed-race African and Latin Americans. Similarly, fewer Hispanic patients responded to IVC than MMF [7]. Further, more patients in the African ancestry group were withdrawn from the study than those in the Caucasian and Asian groups because of adverse events from IVC, while more patients in the Asian subgroup were withdrawn due to side effects caused by MMF [8]. In analyzing B cell targeted therapies clinical trials, patients of African descent responded better to rituximab [9] but were less responsive to belimumab compared to those of other ethnicities, probably due to their higher serum levels of B cell-activating factor of the tumor necrosis factor family [10].
In this review, we hypothesize that the early human migration and human genetic adaptation to various environments, particularly pathogens, contribute to human genetic diversity between races. Moreover, we compiled the current evidence on the ethnic gene diversity related to SLE and its severity.
MAIN SUBJECTS
The race of Homo sapiens: the plausible theory of human genetic diversity between races
1) The early human migration
Humans migrated from East Africa into Northern Europe and Asia between 200,000 and 60,000 years ago (Figure 1). Two conflicting hypotheses have been raised on human evolution: the interbreeding and the replacement theory [11]. According to the replacement theory, Sapiens and other humans, such as Neanderthal and Erectus, would not have had sexual interest in one another. Therefore, their genes should have disappeared following extinction since they had not merged. The replacement theory had been common knowledge until recent decades.
However, the increasing availability of data on DNA in recent years has greatly facilitated the study of human history, showing that Neanderthal-derived DNA accounts for 1% to 4% of modern genomes in people outside sub-Saharan Africa [12]; it is highest in East Asians, intermediate in Europeans, and the lowest in Africans [13]. In conclusion, Sapiens, from African immigrants, bred with Neanderthals so that the two populations merged. Subsequently, today’s Eurasians are a mixture of Sapiens and Neanderthals. As Sapiens reached East Asia, they further interbred with the local Erectus, culminating in the Chinese and Koreans being a mixture of Sapiens, Neanderthals, and Erectus.
2) Human genetic adaptation to various environments, including pathogens
(1) The evolution of immune genes
Human evolution has been affected by different climates, food sources, and pathogens. Subsequently, human skin became lighter, the metabolism adapted to new food sources, and the immune system had to manage different pathogens. Moreover, Homo sapiens have evolved into various subspecies and races [14].
When it comes to pathogens, the first bacteria and virus evolved 3 billion and 1.5 billion years ago, respectively [15]. Pathogens were first observed in Africa 200,000 to 300,000 years ago, spreading to other continents over the last 60,000 to 100,000 years and more rapidly through the emergence of agriculture 10,000 years ago [16].
Infectious diseases are the leading cause of mortality in human history [17]. Since a formal link was established between infectious diseases and natural selection in previous studies showing that thalassemia and sickle cell disease can protect against malaria [18], various evidence has accumulated concerning the genetic determinants of infectious, inflammatory, and autoimmune disorders [19,20].
Furthermore, genome-wide association studies (GWAS) have supported the hypothesis that microbes have significantly affected human evolution [16,20,21].
Recently, statistics of the COVID-19 infection showed racial differences in morbidity and mortality, which are significantly lower in developing countries compared to the developed countries in Europe and North America. A total of 37% of all mortalities during the pandemic occurred in the European Union (EU) [22], even though the population of the EU forms only about 9.78% of the global population. A striking difference in mean per million mortality was reported between Asian and European countries (2.7 vs. 197 deaths per million population, p<0.001). For example, mortality rates were less than 1 death/million in Taiwan, Vietnam, and Thailand compared to 1,112 in Belgium [23].
The antagonistic pleiotropy hypothesis was considered in the pathogenesis of chronic inflammatory diseases, which is supported by the overlap between loci underlying infectious and inflammatory traits [24,25]. Additionally, several pleiotropic variants were reported to confer protection against infectious diseases alongside susceptibility to some chronic inflammatory diseases [26-28]. Indeed, the human leukocyte antigen (HLA) locus is a representative example, whereby variants of the HLA are considered to be under pathogen-driven positive selection [29] and can increase the risk of autoinflammatory diseases, such as ankylosing spondylitis and type 1 diabetes [30-34]. Another example is the common TYK2 P1104A variant, which protects against autoimmune phenotypes (odds ratio [OR]=0.1~0.3) but, in the homozygous state, confers a predisposition to mycobacterium-related infectious diseases (OR >10) [28,35-41]. Thus, selection has likely led to a higher genetic risk of inflammatory gastrointestinal disorders since the Bronze Age. The main risk variants for inflammatory bowel disease are located close to key immunity genes (IRF1, IKZF1, FUT2, and SH2B3), for which monogenic lesions confer susceptibility or resistance to infectious diseases [42-44].
These results suggest that recent temporal changes in pathogen exposure probably caused positive selection to target the regulatory machinery underlying immune cell variation [45,46].
(2) The evolution of major histocompatibility complex polymorphisms
The major histocompatibility complex (MHC) is the most polymorphic gene locus in vertebrates [47]. A co-evolutionary arms race with pathogens is responsible for the high levels of genetic diversity [48]. The HLA system includes MHC class I genes (HLA-A, -B, and -C as classical genes; HLA-E, -F, and -G as nonclassical genes), MHC class II genes (HLA-DP, -DQ, and -DR as classical genes; HLA-DM and -DO as non-classical genes), and MHC class III genes (complement and cytokine genes). HLA-DRB1 is the most variable region in HLA class II genes, with over 2,000 registered alleles [49,50] since HLA-DRA is basically invariant. HLA-DRB1 shows the strongest general signature of selection among HLA class II loci [51] and has evolved and diversified very rapidly [52] after ancestral human migration owing to encounters with many novel pathogens [53]. HLA-DRB1 is more highly expressed on the cell surface than other HLA class II molecules [54].
Another significant variation in MHC molecules is the epitope repertoire size, with an up to 16-fold variation in the number of bound peptides [55]. A group of promiscuous MHC molecules that enhance the immune response against a broader range of pathogens are called generalists, in contrast to specialists, which exhibit a response against fewer pathogens [56]. A tiny difference in the amino acid sequence shows massive promiscuity range differences. Even though the HLA-DRB1*13 group alleles present an amino acid sequence identity to each other of over 98%, the promiscuity levels vary 57-fold [57]. High pathogen diversity increases the MHC diversity in relation to their average genomic diversity [29], and the diversity of MHC class II molecules is found in the molecular positions that define promiscuity level, which is under positive selection [57]. Interestingly, HLA-DRB1*12:02 has the highest promiscuity level among HLA-DRB1 alleles in Southeast Asia, which protects against pulmonary tuberculosis and typhoid fever—endemic diseases in this area [58]. Therefore, the high allele frequency of DRB1*12:02 reflects the pathogen-driven selection that occurred during the migration of Mongolians to South China [59].
3) Space invaders: endogenous retroviral sequences in systemic lupus erythematosus
Human endogenous retroviruses (HERVs) are viral elements in the human genome that could be derived from retroviruses. Most HERVs transitioned into the human genome around 25 million years ago [60], and about 8% of the human genome consists of retroelements containing long terminal repeats, a group that includes endogenous retroviruses [61]. Some HERVs are still active, and various factors, including ultraviolet radiation, inflammatory cytokines, steroid hormones, and exogenous virus products, could reactivate their expression [62]. HERVs have been considered to induce multiple autoimmune diseases [63]. Currently, 200 endogenous retroviral sequences have been reported, and HRES-1, HERV-E 4-1, HERV-K10, and HERV-K18 HERVs are related to the pathogenesis of SLE [64-66]. Present hypotheses consider that autoantibody production occurs via molecular mimicry, stimulation of intracellular sensor molecules by viral nucleic acids, and epigenetic regulation of host genes mechanisms [67,68]. Antibodies against HRES-1, which is an endogenous retroviral element-encoded nuclear protein autoantigen, were higher in patients with SLE compared to control subjects (52% vs. 3%) [69]. The molecular mimicry hypothesis has also been raised since cross-reactivity is reported between HERV proteins and self-antigens [70].
The race of Homo sapiens lupus: the genetic architecture of systemic lupus erythematosus
The pathogenesis of SLE is influenced by a strong genetic contribution, with a 66% of heritability [71]. The SLE has a significant genetic predisposition, influenced by both ancestry-dependent and ancestry-independent factors. Over the past decade, over 100 susceptibility loci have been identified by GWAS and reported, mostly in European and Asian populations [59,72,73]. The polygenic etiology of SLE is supported by >170 SLE-risk-associated genomic loci characterized by common variants with modest effect sizes identified through GWAS or candidate-gene approaches in multiple ethnic groups [74-77].
1) Human leukocyte antigen loci associated with systemic lupus erythematosus
HLA-DRB1 is widely known for its pivotal role in regulating immune tolerance to self-antigens. The HLA-DRB1 allele frequencies vary in the general population of different ethnic groups [78]. For example, HLA-DRB1*03:01, *07:01, and *15:01 were predominantly identified in Europeans; *03:01, *07:01, and *11:01 in African Americans; *15:01 and *09:01 in Asians; and *09:01 and *04:05 in Koreans (Table 1).
The primary SLE association signal has been consistently identified within the HLA-DRB1 gene in multiple ancestral populations [75-77,79]. These findings underscore the significance of the HLA-DRB1-mediated immune response in developing SLE. Multiple ancestral studies revealed that the HLA-DRB1*15:01 and *03:01 alleles were the most significant SLE-risk factors within the HLA class II region, which is shared across multiple ancestries, including African American, Asian, and European [25,74,75]. HLA-DRB1*03:01 is the largest SLE-risk allele in European ancestries [74], although it is not common enough to detect its association in East Asian SLE cohorts [25,76,77,80]. European patients with SLE were linked to HLA-DRB1*03:01 and *08:01 [74]. In contrast, Asian patients with SLE possessed more copies of the HLA-DRB1*08:03, *07:01, and *09:01 alleles than the healthy controls [76].
Identifying the genetic variants responsible for SLE development and its clinical manifestations has been challenging. The HLA-DRB1*15:01 allele was positively associated with many clinical sub-phenotypes of SLE [76]. In addition, the *09:01, *08:03, or *07:01 alleles were linked to SLE risk and significantly affected more diverse clinical manifestations [76]. Notably, individuals carrying double copies of the HLA-DRB1 risk alleles exhibited an elevated risk of producing anti-Sm antibodies [76].
Recently, a detailed analysis of HLA amino acid residues provided an improved understanding of the specific HLA associations based on ethnicity. Specifically, the amino acid positions 11–13–26 in HLA-DRB1 at the epitope-binding groove of the HLA-DR molecules were most significantly associated with increased susceptibility to SLE by Asian and European populations [75]. The two well-known SLE-risk HLA-DRB1 alleles *15:01 and *03:01 belonged to their respective SLE-risk haplotypes, Pro–Arg–Phe and Ser–Ser–Tyr, at amino acid positions 11, 13, and 26. The haplotype comprising Asp–Phe–Tyr at amino acid positions 11, 13, and 26, respectively, is commonly found among Asian populations, while the Ser–Ser–Tyr haplotype is more common among Europeans (Table 2). The amino acid positions 11 and 13 in HLA-DRB1 have been reported to be linked to SLE susceptibility and the production of autoantibodies, thereby driving the pathogenesis of LN [75,81]. These amino acid positions within the epitope-binding groove of HLA molecules contribute to the risk of developing SLE.
2) Non-human leukocyte antigen loci associated with systemic lupus erythematosus
The reported non-HLA loci associated with SLE have been identified in African American, Asian, European, and shared ethnic groups (Supplementary Table 1), which may regulate gene expression and contribute to SLE development.
We investigated ancestry-driven key signaling pathways in African American, Asian, and European populations by analyzing reported non-HLA single-nucleotide polymorphism (SNP)-associated genes using enrichr (https://maayanlab.cloud/Enrichr/#libraries) (Supplementary Table 1 and Figure 2).
International collaboration in diverse ancestral populations has facilitated the identification of SLE-risk loci and revealed the heterogeneity of SLE variants among ancestries.
Asian-associated genes were dominated by the functional category for type-I interferon-stimulated genes and the janus kinase–signal transducers and activators of transcription (JAK–STAT) signaling pathway (Figure 2), along with the top pathways determined by Gene Ontogeny terms for each gene list. Europeans are associated with the type-II interferon-stimulated genes and IL-12 and IL-17 signaling pathways, while Africans are associated with the B cell and IL-12 signaling pathways.
The pathogenesis of SLE is linked to diverse immunological pathways of SLE-risk SNP-associated genes. Thus, clinical trials using many drugs against different targets, such as B cell (CD20, CD22), type-I interferons, JAK, tyrosine kinases, interleukin (IL)-12 and IL-23, or plasmacytoid dendritic cell receptors have recently been conducted (Table 3) [82-89]. Ethnic diversity in response to each treatment should be further investigated in these trials.
3) Weighted genetic risk score in different ethnicities
Each SLE patient presents with significantly different symptoms, prognoses, and mortality rates. Therefore, it is important to assess individual genetic susceptibility using a cumulative weighted genetic risk score (wGRS), defined as the sum of the number of risk alleles at various loci in an individual, weighted by their odds ratio.
Analyzing wGRS across five ethnic populations revealed that the higher wGRS and SLE risk allele frequencies showed a greater SLE prevalence (Supplementary Table 2). The populations ranked from the lowest to highest for the average wGRS were Europeans, Amerindians, South Asians, East Asians, and Africans [79]. A higher wGRS for SLE was observed more often in childhood-onset SLE than in adult-onset SLE [90,91].
In addition, a high wGRS based on 57 SLE risk loci was significantly correlated with a higher risk of renal disorder, end-stage renal disease, and mortality in a Caucasian population [92].
A recent study of wGRS using 112 non-HLA SNPs and HLA-DRB1 amino acid haplotypes in Korean patients with SLE revealed that a higher wGRS was significantly correlated with higher clinical manifestations of SLE [93]. Notably, the highest quartile of wGRS was associated with the highest risk of LN and the production of anti-Sm, regardless of onset age. Furthermore, an increasing wGRS significantly impacted the development of proliferative LN (class III or IV) or membranous LN (class V) [93]. The genetic risk load of SLE significantly influenced the development of diverse clinical manifestations and LN; thus, genetic profiling using wGRS could enable personalized management by predicting the clinical course of SLE.
CONCLUSION
The future of Homo sapiens lupus
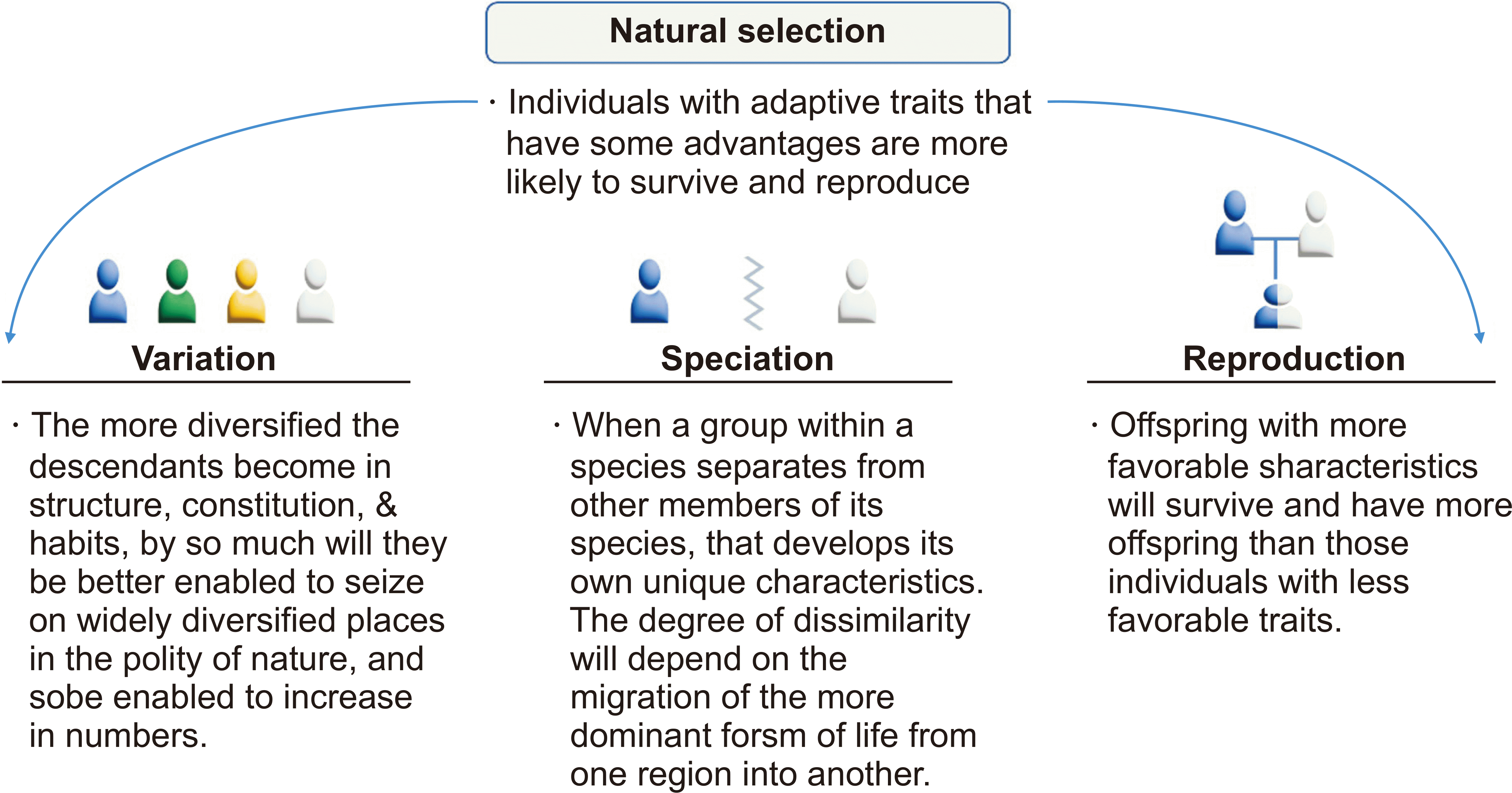
According to the natural selection theory, individuals who are more adapted to their environment are more likely to survive and pass on genes important to their survival (Figure 3) [94]. For this theory to be correct, genes associated with lupus should have been deleted. Furthermore, if genes associated with lupus have maintained under pathogen-driven positive selection, the risk of infection should be lower in lupus.
The theory of geographic distribution would make lupus patients different by region but similar in the region over time, meaning we could still manage the disease using designated medicines for a specific race or an approved region of the globe. However, these circumstances would not happen because genetic variation currently occurs more between individuals than ethnic groups.
The genetic risk load and clinical outcomes across different ethnicities could improve SLE management based on individual genetic risk profiles.
According to variation theory, lupus patients will become more genetically diverse in the future, comprehensive genetic studies should be continuously conducted to find out better targets, which will pave the way for personalized treatment for SLE patients.
SUPPLEMENTARY DATA
Supplementary data can be found with this article online at https://doi.org/10.4078/jrd.2024.0051
 XML Download
XML Download