

 PDF
PDF Citation
Citation Print
Print



Introduction
Huntington’s disease (HD) is a rare hereditary neurodegenerative condition marked by the progressive emergence of physical limitations, psychiatric symptoms, and cognitive deterioration [1]. The global rate of HD is about 2.7 cases per 100,000 [2]. Nevertheless, it is widely acknowledged that there is geographical variation in its incidence. Compared to Asian countries, the condition is more prevalent in Western nations, including the Australia, United States, Canada, and the United Kingdom [3, 4]. HD is a hereditary condition passed down in an autosomal dominant manner and arises due to the expansion of a trinucleotide sequence, known as cytosine-adenine-guanine (CAG), inside the coding domain of the gene associated with HD. This gene is responsible for the production of the Huntingtin (HTT) protein, found in various tissues, including the central nervous system (CNS). Although its precise actions remain incompletely understood, it has been postulated to fulfil crucial functions in several cellular processes, including vesicle transport, protein trafficking, and selective autophagy [5, 6]. When repetitions of CAG trinucleotide sequence exceed the typical range of 6–26, it exhibits instability and has the potential to expand in later generations, particularly when transmitted paternally. The popularly accepted threshold for the development of HD is often regarded as 36 repetitions or more, however complete penetrance is not shown until there are at least 40 repeats present [7, 8]. The symptoms of HD often manifest in people aged among 30–50 years and can be categorized into three primary domains: motor, psychiatric, and cognitive [9, 10]. Chorea, a prominent motor manifestation of HD, is characterized by transient and involuntary movements that often impact the trunk, facial region, and upper extremities. Additional Motor/physical symptoms are the emergence of bradykinesia, dystonia, hyperreflexia, and deceleration of ocular saccades [11]. HD does not have a definitive cure, but drug therapy, physiotherapy, occupational therapy and speech therapy are also effective in reducing the complications caused by this disease. Recently, some neuroprotective substances are used for treatment HD, such as Apelin, Apelin, which is a neuropeptide with bioactive properties, has been shown to functions as an ligand for the apelin receptor (APR) [12]. Widely recognized is the fact that proteases play a significant role in cleaving prepro-Apelin, contributing to the generation of physiologically-active Apelin peptide, including Apelin-12, -13, -17, and -36 [13]. Within this set of peptides, Apelin-13 exhibits the highest level of biological activity in comparison to the others [14]. Comprising 380 amino acids, APR stands as a prototypical G protein-coupled receptor. It exhibits significant sequence similarity, with around 30%–40% amino acid sequence of the angiotensin II receptor type 1 [15]. APR has been identified in different parts of the CNS like cerebral cortex, thalamus, hypothalamus, midbrain, reticular formation, basal ganglia, glial cells and white matter [16]. Previous research has shown a significant association between Apelin and the advancement of HD. A recent study illustrated that that Apelin facilitates the upregulation and phosphorylation of cytoskeletal components via the regulation of PI3K/Akt and MAPK/ERK signaling pathways, therefore, facilitating the microtubule-mediated transport [17]. Additionally, the administration of insulin-like growth factor 1 (IGF1) can promote microtubule transport and metabolic function. This improvement subsequently leads to the elimination of HTT protein aggregates, and then improve mitochondria functionality, alleviation of motor irregularities, and increased endurance and survival. Interestingly, the upregulation of Apelin expression has been seen in response to IGF1 [18]. Hence, it is plausible that Apelin has a significant role in supporting the preservation of axons and dendrites by controlling the process of cytoskeleton remodeling. Recent studies have suggested that Apelin-13 may have potential therapeutic benefits for HD. Apelin-13 has been shown to shield neural cell from cell death caused by mutant HTT and reduce inflammation in the brain [18]. Although now it is used for the treatment of neurodegenerative diseases from mesenchymal stem cells and neural stem cells and their differentiation, but it seems that apelin can be a suitable option for treatment [19]. Overall, the association between Apelin-13 and HD seems to be multifaceted, with Apelin-13 potentially playing a protective role against the neurodegenerative effects of mutant HTT in HD [12-15]. Consequently, we intended to assess the impact of Apelin-13 HD rat models induced by 3-NP.
Go to : 
Materials and Methods
Animals and HD model
Thirty male’s adult Wistar rats with weights ranging from 200–220 g were obtained from the Laboratory Animal Center of Shahid Beheshti University of Medical Sciences (Tehran, Iran). The animal experiment conducted received approval from the Shahid Beheshti University’s Medical Research Ethics Committee (IR.SBMU.AEC.1401.054). The rats were placed in cages, maintaining constant humidity and temperature at 22°C, following 12-hour light and 12-hour dark cycle. All rats were randomly divided into three groups as follows: control (n=10), HD (n=10), and HD+Apelin-13 (n=10). To initiate the development of HD in these animals, all rats in the HD and HD+Apelin-13 (Sigma-Aldrich) groups were administered intraperitoneal injections of 3-NP for duration of five consecutive days (30 mg/kg, Sigma-Aldrich).
Rotarod test for evaluating motor coordination
One week before the initiation of the experiment, daily sessions of the training trial were conducted. The rotarod performance assessment took place consistently over a period of four weeks subsequent to the final administration of 3-NP on a designated day each week. Throughout the trial, animals were positioned on the accelerating cylinder, ranging 4–40 rpm, with individual testing session lasting 300 seconds. The trial was halted if the rat dislodged from the rods, clung to the device, or completed two consecutive turns. Ultimately, the recorded parameter was the maximum duration that each rat successfully performed the task.
Locomotion tracking apparatus: open field and elevated plus maze
To evaluate both locomotor activity levels and anxiety across all experimental groups, an open field test was performed subsequent to the final injection of 3-NP. This test involved placing the animals into a square field measuring 90 cm in both height and length.
At the start of the test, rat was located in one angle of the apparatus. The camera (Ethovision; Noldus), situated on the ceiling above the open field arena, automatically recorded the total distance covered by the animals. Ethovision software (version 7) facilitated data acquisition, with recording concluding after 5 minutes, after which the rats were all returned back to designated cages. Additionally, the field underwent cleaning and drying after each trial. To minimize the potential impact of unexpected disruptive environmental factors, behavioral tests were carried out in a serene, enclosed setting. The animals’ anxiety levels were expressed by determining the duration they spent in the center or corner of the apparatus. The elevated plus maze, which is a common behavioral test in rats can be used to measure anxiety-like behavior. The elevated plus maze comprises a raised structure with two open arms and two enclosed arms. The rat is introduced into the central area of the maze and permitted unrestricted exploration for a set period of time, typically around 5–10 minutes.
Anxiety-like behavior is measured based on the rat’s tendency to avoid the open arms, which are more exposed and therefore perceived as riskier or threatening. Rats that exhibit higher levels of anxiety will spend more time in the enclosed arms and less time in the open arms.
Electromyography after sciatic nerve stimulation and compound muscle action potential recording
Rats were anesthetized via intraperitoneal injection of xylazine (at a dosage of 8 milligrams per kilogram) as well as ketamine hydrochloride (at a dosage of 60 milligrams per kilogram). Initially, the rat’s right hind limb underwent shaving and thoroughly cleansing with Betadine. A longitudinal incision of 3 centimeters was created on the posterior part of the thighs, stretching from the rat’s greater trochanter site to the knee. Subsequently, in order to clearly expose and activate the sciatic nerve within the gastrocnemius muscle, the dissection focused on isolating the muscles extending from the rat’s gluteus maximus muscle to the biceps femoris muscle. Careful application of forceps avoided any harm to the nerve exposed, making it accessible. The sciatic nerve was then carefully isolated from the surrounding connective tissue, and electrodes for stimulation were positioned beneath it. Two monopolar electromyography (EMG) needle electrodes were positioned parallel to each other, maintaining a distance of 7 mm, as stimulation electrodes. The electrodes used for recording were insulated, except for the distal part, the sciatic nerve was subsequently subjected to stimulation, with an amplitude of 1 A, a frequency of 0.2 Hz, and lasting for a duration of 100 seconds. Concurrently, the compound muscle action potential (CMAP) assessed the overall electrical activity of gastrocnemius muscle from the stimulated limb. The CMAP parameter, specifically latency, was considered. Following the procedure, the incision was stitched closed, and the rats were then placed back to their designated cages, being monitored until they were completely recovered.
Tissues preparation for striatal volume estimation
The brain tissue specimens underwent fixation within formalin (10%) for a period of seven days. Subsequently, sequential coronal sections with a thickness of 10 µm were created and underwent hematoxylin and eosin (H&E) staining. Systematic sampling technique was used to randomly choose sample data (20 sections) for each individual animal that represents a population.
Quantifying the total count of neuron and glial
Cavalieri’s principle was used to evaluate total striatal volume. The density of neurons and glial cells (Nv) in the striatum was determined by the optical dissector technique, as per the following equation:
Nv=∑Q/∑P×h×a/f×t/BA:
In the given equation, ΣQ represents the total nuclei number, h denotes the dissector’s height, Σp stands for the overall count of frames, a/f represents the area of each frame, BA is the tissue section thickness, and t signifies the actual thickness of section [20].
Immunohistochemistry
Rats underwent anesthesia using chloral hydrate and subsequently underwent trans-cardial perfusion with saline, which is followed by perfusion with a fixative consisting of 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS) (Sigma-Aldrich). Subsequently, the rat’s brains were extracted and immersed in the fixation solution with the same formula for a duration of 24 hours. The tissues underwent dehydration via multiple ethanol baths of varying concentrations to eliminate water content and were then embedded in paraffin waxes. The microtome was utilized to cut tissues into sections of 5 μm thickness. Following this, the tissue sections embedded in paraffin and fixed with formalin were then treated to remove the paraffin and hydrated. For the purpose of immunohistochemistry, the primary antibody, were diluted within PBS buffer (Sigma-Aldrich) having 0.3% Triton X-100 (Sigma-Aldrich) and supplemented 1% bovine serum albumin (Merck). Tissues were subjected to overnight primary antibody incubation at 4°C against GFAP (Abcam) and inflammatory factor (Iba-1, Abcam) (dilution concentrations 1:100). In the subsequent step, the sections underwent incubation with the avidin-biotin complex, followed by incubation with 3,3-diaminobenzidine tetrahydrochloride (0.05%) and hydrogen peroxide (0.03%) within 0.05 M Tris-buffer (pH 7.6). The positive areas associated with GFAP and Iba-1 antibodies were quantified. By using Image J software, positive cells in groups were measured. The threshold was adjusted, and examination was done for totally selected images. The mean±SD of the final data was described [19].
Reactive oxygen species assay
The measurement of reactive oxygen species (ROS) followed the procedures outlined by Keston and Brandt (1965) with some level of optimization. After brain dissection, in order to achieve a 5 mg/ml tissue concentration, the tissue solution was further diluted at a ratio of 1:10 in the buffer. Subsequently, the brain solution was transferred into 24-well plates (0.45 ml/well) and left at 25°C for a period of 5 minutes. During this period, with the aim of incorporating 2’-7’-dichlorodihydrofluorescein diacetate (DCFH-DA) (Sigma-Aldrich) into any membrane-bound vesicles, 5 ml of DCFH-DA (final concentration of 10 mM) was pipetted into individual well, and subseuqntly plates were left at 25°C for 15 minutes. Esterases cleaved the diacetate group, and subsequent to the preincubation, 50 ml of Fe was pipetetd to the wells. Following a 30-minute incubation, DCFH underwent oxidation, leading to the generation of a fluorescent product, dichlorofluorescein, which was subsequently measured by fluorescence spectrophotometer. The concentration of proteins in tissue solution was assessed via Bradford’s method using a commercial kit [21].
Measurement of reduced glutathione content and glutathione disulfide
The working reagent, 5,5’-dithiobis (DTNB) (Sigma-Aldrich), composed of 50 μl of DTNB, 100 μl of Tris (Sigma-Aldrich), and 840 μl of distilled water, was quantified using a spectrophotometer. Subsequently, 10 μl of tissue-lysed buffer was combined with 990 μl of the DTNB reagent, thoroughly mixed, and left to incubate at room temperature for 5 minutes. Concentration of glutathione (GSH) were quantified in microliters. DTNB, known as Ellman’s reagent, was utilized for the detection of thiol compounds, enhancing the sensitivity of total GSH detection through a recycling reaction. The yellow-colored product, 2-nitro-5-thiobenzoic acid, generated from the reaction, enabled the quantification of GSH absorption in a sample by measuring absorbance at 412 nm [21].
Data analysis
Numerical results are presented as mean±standard error of the mean. Variations among groups were examined through one-way ANOVA, following by Tukey’s test. A significance level of P<0.05 was considered statistically significant.
Go to :
Results
Rotarod result
The rotarod test was performed to evaluate the effect of Apelin-13 on motor coordination after induction of HD in rats. The motor coordination was significantly decreased in the HD group in comparison with the control group (P<0.001). In contrast, the rotarod scores (latency to fall) increased significantly in the HD+Apelin group in comparison with the HD group (P<0.001). Indeed, after the intraperitoneal administration of Apelin-13, motor coordination significantly improved over a period of 4 weeks, with notable enhancement observed, particularly during the second week (Fig. 1).
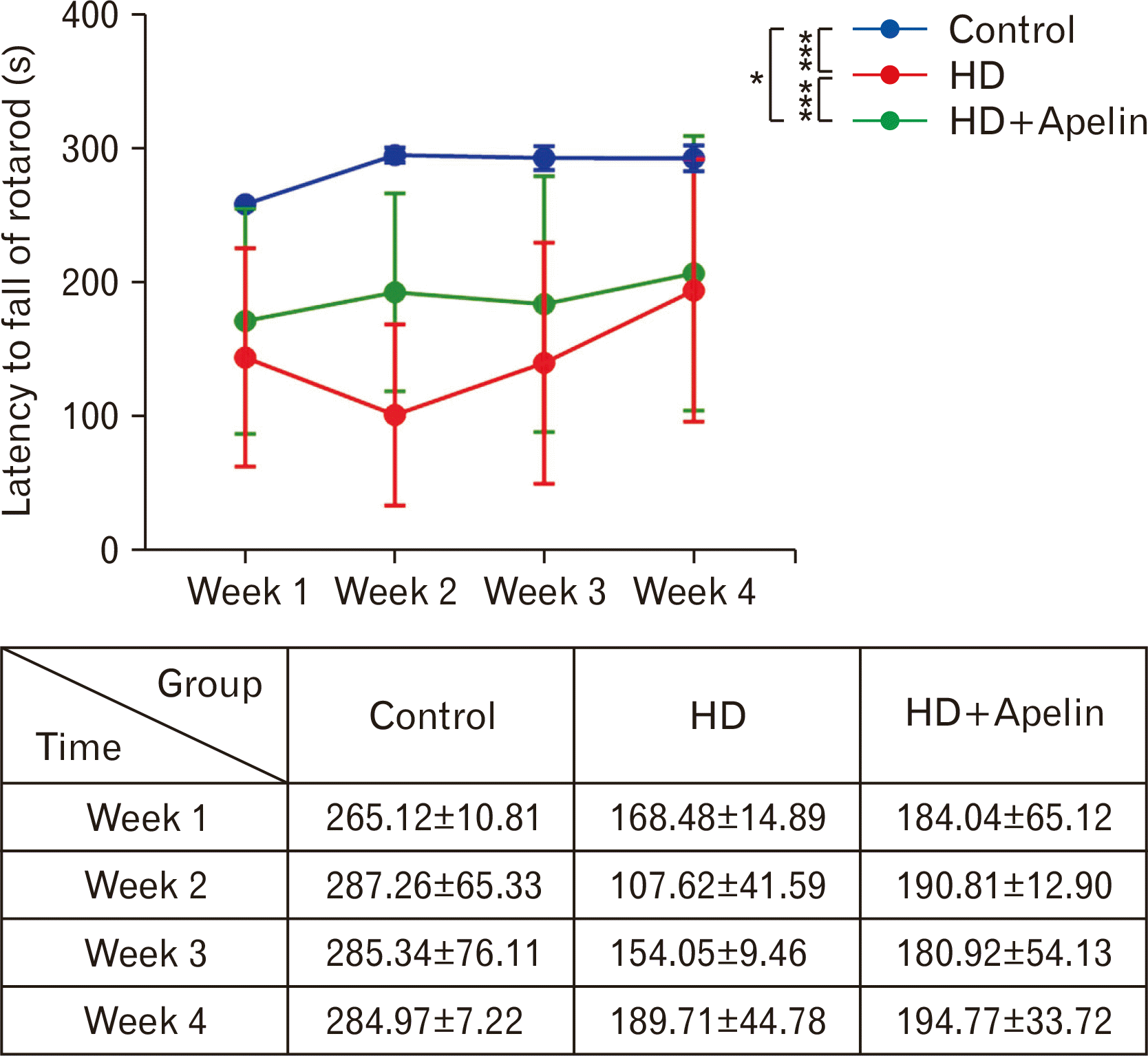 | Fig. 1Locomotion evaluation using rotarod test. As indicated in this figure, Apelin-13 administration following 3-NP administration resulted in increased latency to fall throughout all weeks compared to the Huntington’s disease (HD) group (***P<0.001), also a significant diffidence between control and HD+Apelin group (*P<0.05).
|
EMG result
To assess the impact of Apelin-13 on muscle activity, EMG was conducted. The EMG latency increased in the HD group compared to the control group (P<0.001). However, following Apelin-13 injection, the latency decreased compared to the HD group (P<0.05). Intraperitoneal management of Apelin-13 demonstrated a beneficial effect on muscle activity recovery by reducing EMG latency (Fig. 2).
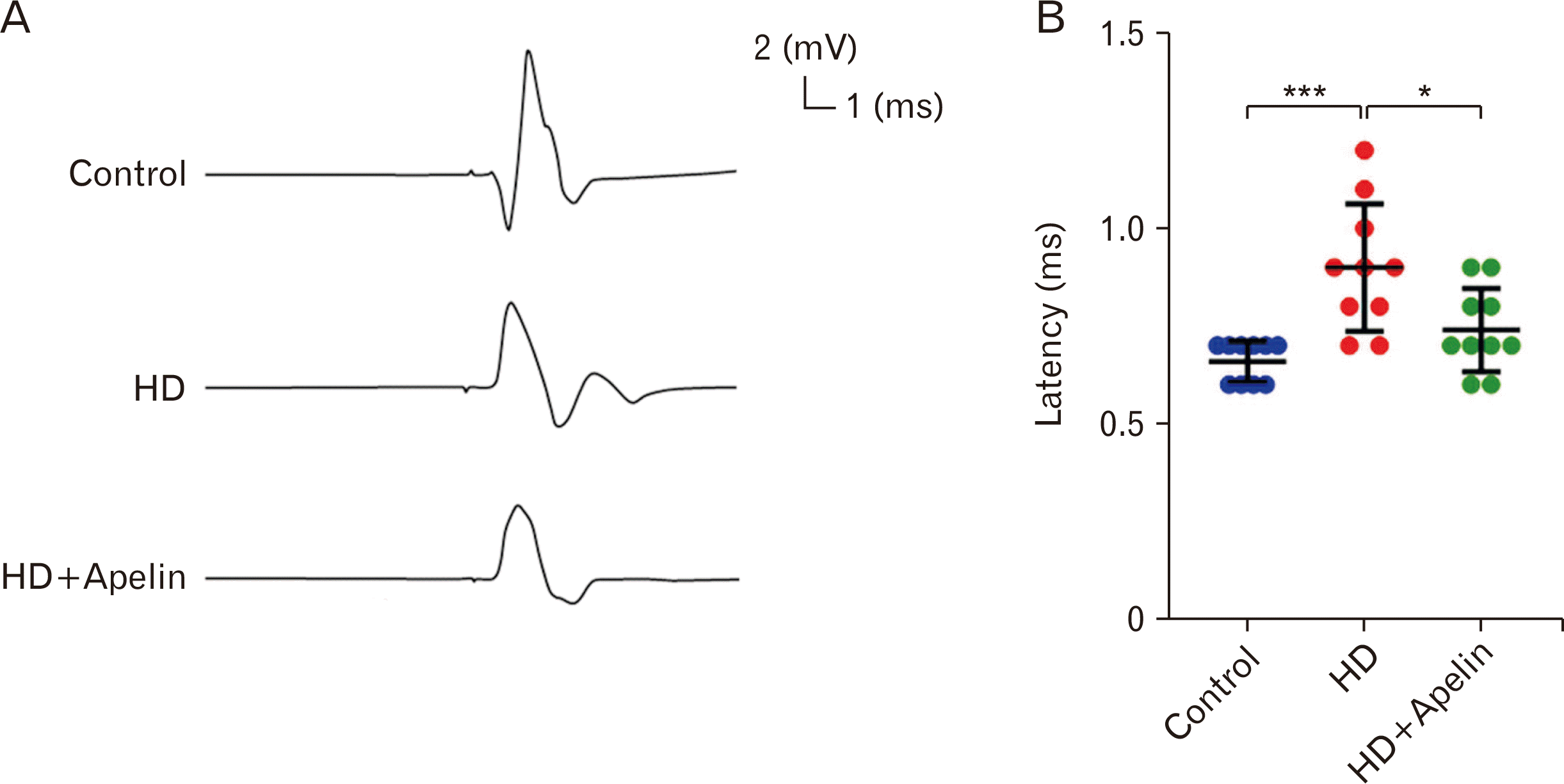 | Fig. 2(A) Apelin-13 treatment improved muscle electromyography (EMG) activity in Huntington’s disease (HD) group. EMG was assessed in all experimental groups, as illustrated in this figure. (B) The EMG latency was sensibility expanded after 3-NP administration (***P<0.001), though it restored in Apelin-13 rats (*P<0.5).
|
Open field and elevated plus maze test
The elevated plus maze test serves as a tool to evaluate anxiety-like behavior in animal models of the disease. Result showed that rats in HD group showed that increased anxiety-like behavior open arm time (OAT%) and open arm entry (OAE) on the elevated plus maze in comparison with control group (P<0.001). This is believed to stem from dysfunction in brain areas responsible for modulating anxiety and fear responses, notably the amygdala and prefrontal cortex, both of which are impacted by HD. The open field test is usually used for behavioral studies, motor deficits and anxiety and depression in rats. This test is very useful and effective in studying and evaluating the effects of anti-anxiety and depression drugs, and the reactions of motor organs to substances such as drugs, as well as behavioral reactions. The result of open field showed that 3-NP administration significant decrease in distance travelled and time in center (P<0.001) (Fig. 3).
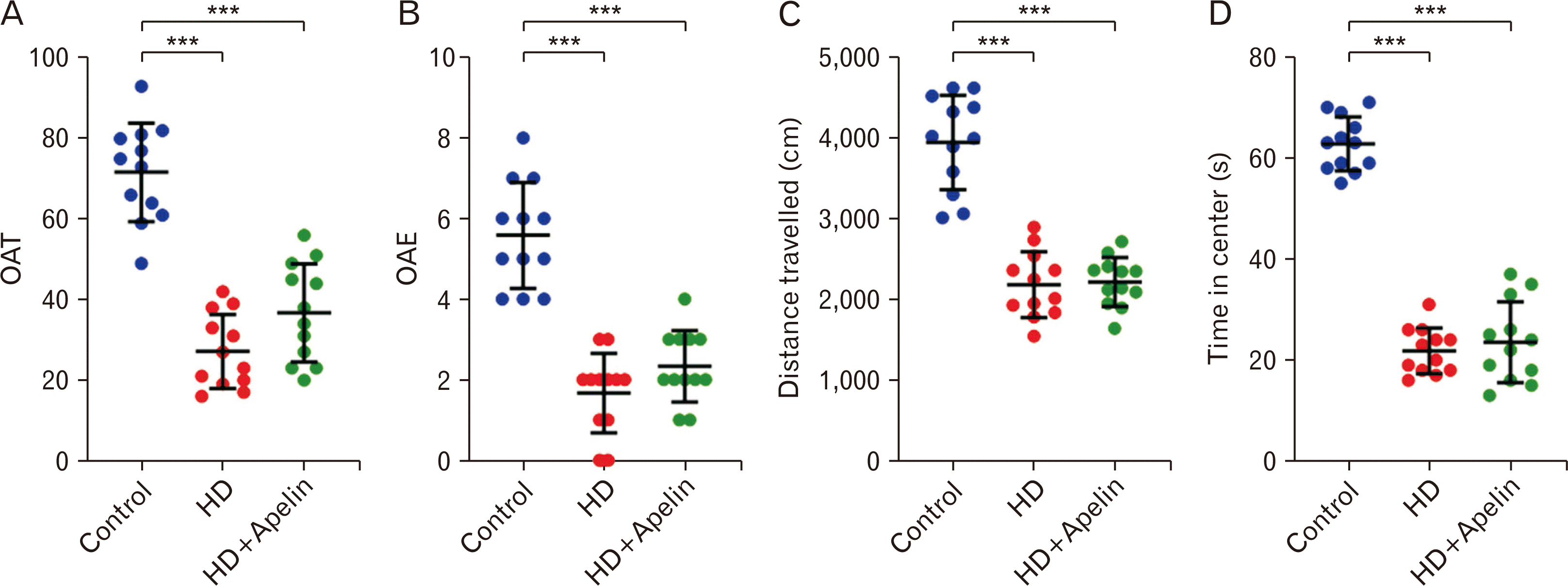 | Fig. 3Elevated plus maze test (A, B) and open field (C, D) result. The results of these test showed that 3-NP administration significant decrease in OAT and OAE (***P<0.001). The elevated plus maze test in Huntington’s disease (HD) group exhibit increased anxiety-like behavior (decreased distance travelled and time in center) in comparison with control group (***P<0.001). Open field result showed the Apelin-13 decrease distance travelled and time in center comparison to the control group (***P<0.001).
|
Reactive oxygen species, glutathione, glutathione disulfide result
According to the study results, 3-NP injections led to a significant increase in ROS and glutathione disulfide (GSSG) concentration in the striatum compared to the control group (P<0.001). However, following HD, the administration of Apelin-13 resulted in a notable decrease in ROS and GSSG concentration. The graphs showed that HD group decreased GSH concentration compared with control group (P<0.001). In contrast, the result indicated that GSH concentration led to significant increase in HD+Apelin-13 group in comparison with the HD group. This test indicated the antioxidant effects of Apelin-13 following HD induced (Fig. 4).
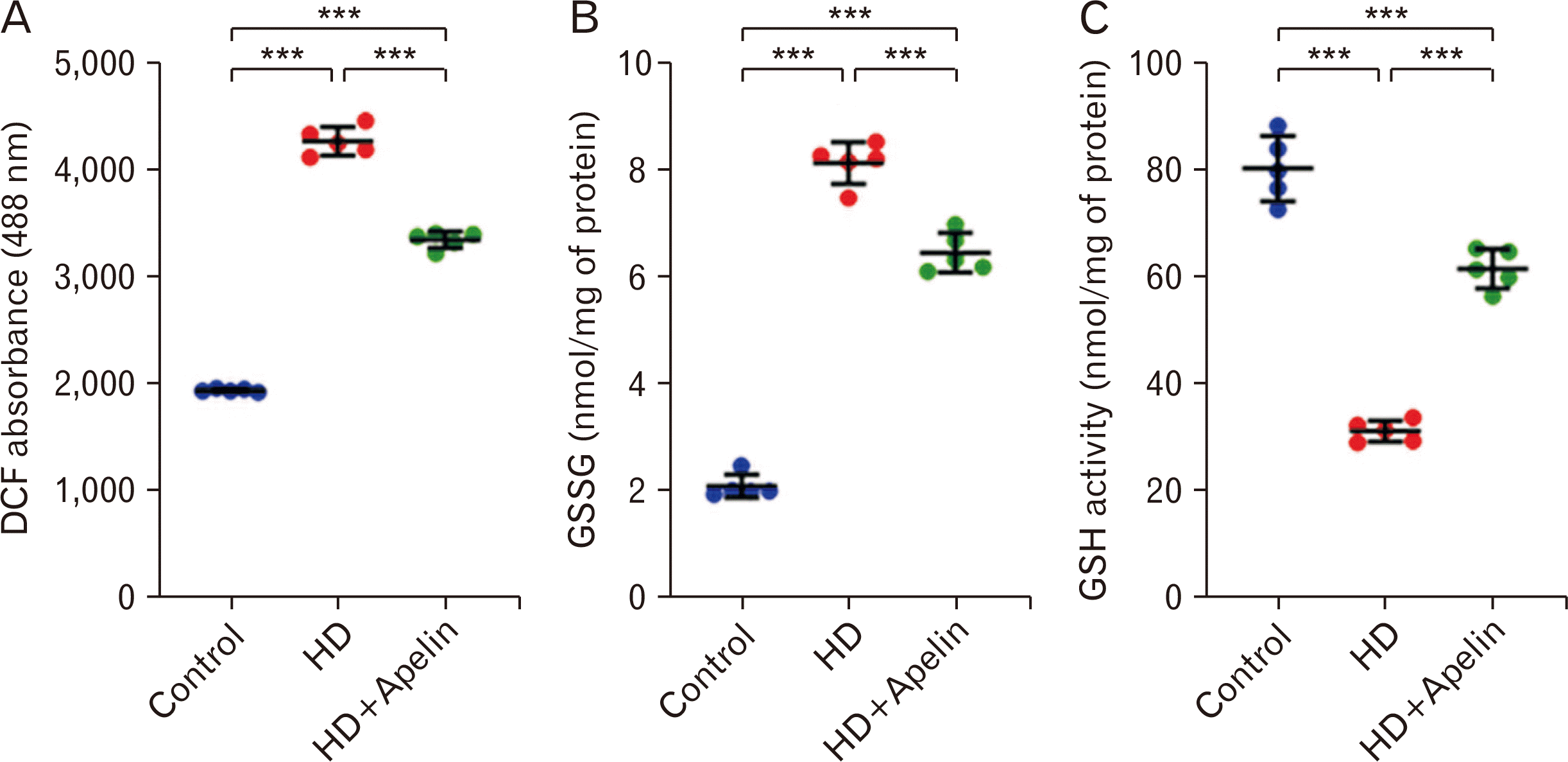 | Fig. 4The results revealed that the levels of reactive oxygen species (A) and glutathione disulfide (GSSG) (B) were elevated in the Huntington’s disease (HD) group compared to the control group. However, following treatment with Apelin-13, there was a significant decrease in the levels of these factors (***P<0.001). Conversely, the amount of glutathione (GSH) (C) decreased in the HD group compared to the HD+Apelin-13 group (***P<0.001). Also we observed significance difference between control and HD+Apelin group (***P<0.001). DCF, dichlorofluorescein.
|
Histology and stereology
According to the stereological analysis, the numerical densities of glial and neurons within the striatum were assessed to gauge the neuroprotective impact of Apelin-13 on neuronal density. The outcomes revealed a significant increase in the numerical density of glial cells in the striatum and a considerable decrease in neurons after 3-NP injections. Conversely, there was a notable decrease in the total count of glial cells and a significant increase in neurons in the HD+Apelin-13 group (Fig. 5).
 | Fig. 5Histological images (upper row) and stereological analysis (lower row) of neuronal and glial cells in all groups (H&E staining). As illustrated in there was significant difference between the quantity of neural and glial cells before and after used of Apelin-13, which neural cells notably increased in the HD+Apelin group in copmarison with HD group (**P<0.01) and glial cells remarkably decreased after administered of Apelin-13 (***P<0.001). Also we observed significance difference between control and HD+Apelin group (*P<0.05). Control group: A, B, C. Huntington’s disease (HD) group: D, E, F. HD+Apelin group: G, H, I.
|
Immunohistochemistry (GFAP, Iba-1)
GFAP, a type-III intermediate filament, serves as a cell-specific marker distinguishing astrocytes from other glial cells. On the other hand, Iba-1 acts as a calcium-binding protein specific to microglia/macrophages. A key indicator of the brain’s response in inflammatory conditions is the activation and proliferation of microglia, referred to as microgliosis. Immunohistochemistry results shown a significant increase in the concentration of GFAP and Iba-1-positive cells in the striatum of the HD group compared to the control group (P<0.001). However, there was a notable decrease in the expression of GFAP and Iba-1 in the HD+Apelin-13 group compared to the HD group (P<0.001), indicating reductions in astrogliosis and microgliosis (Figs. 6, 7).
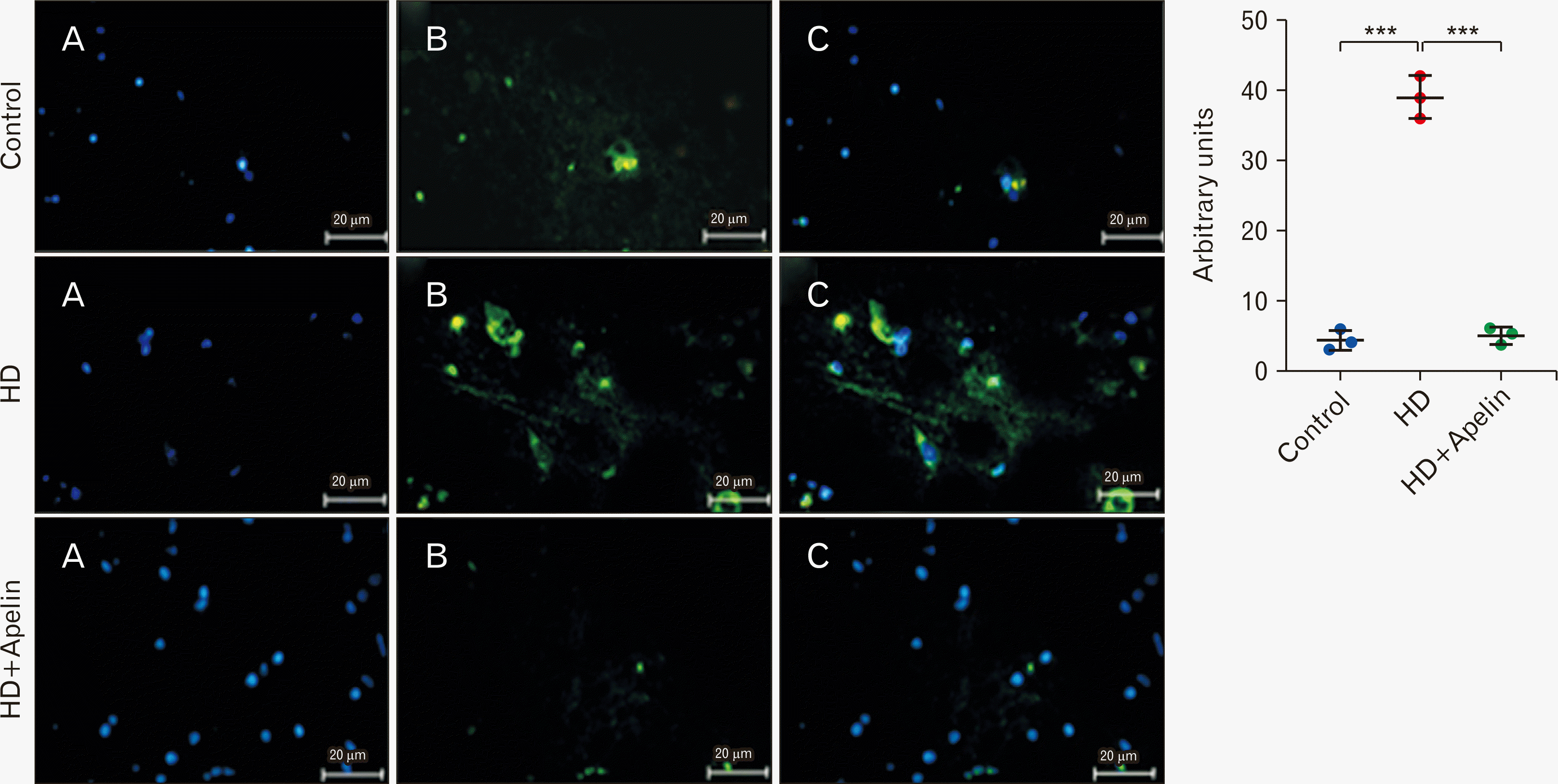
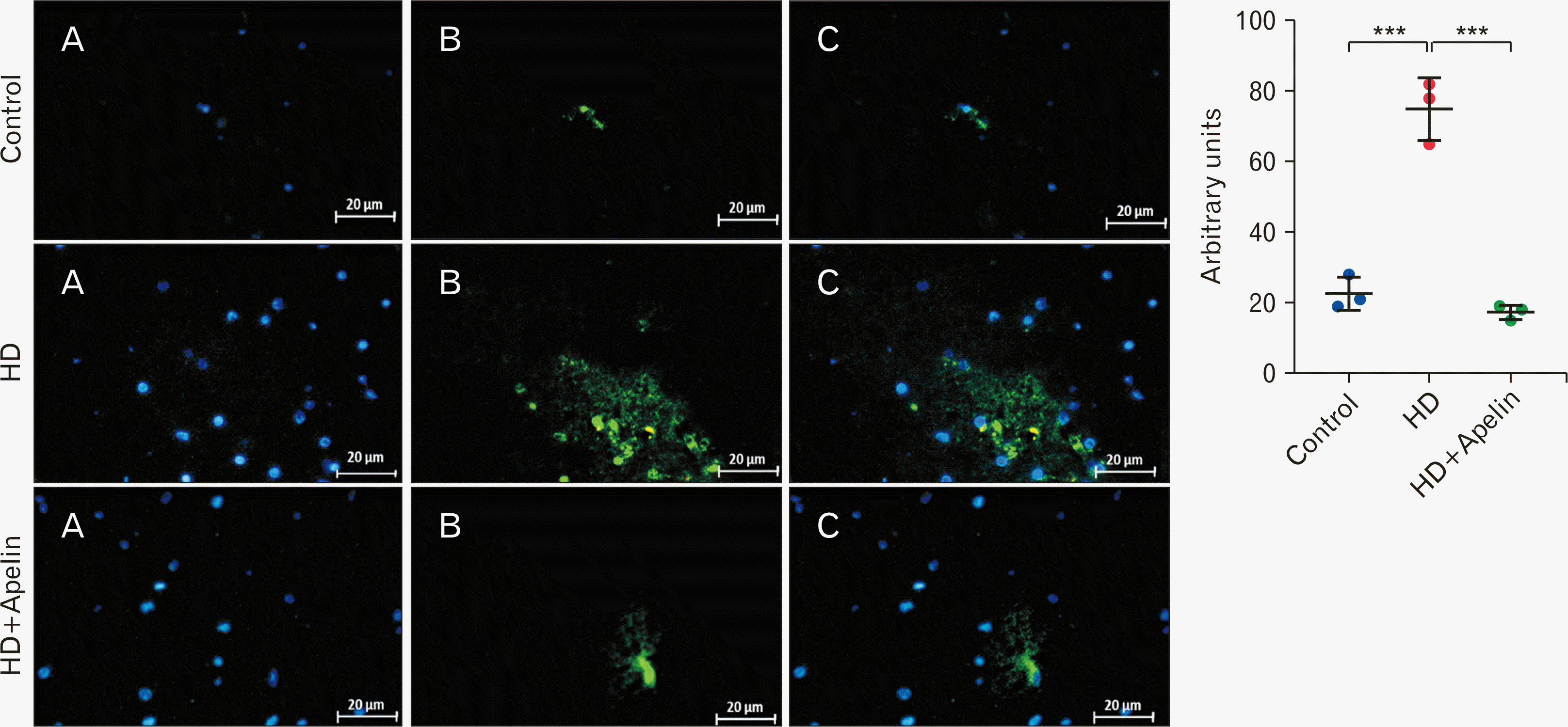
Go to :
Discussion
The result of this research showed that Apelin-13 exhibits a positive impact on the therapy of HD. Furthermore, it has been observed that various tissues of patients with HD, including plasma, postmortem brain tissue, lymphoblasts, and cerebrospinal fluid, demonstrate increased oxidative damage [22, 23]. It seems that Apelin-13, causes recovery in motor function with neuroprotective effects, and prevents cell death. In addition, the analysis of the EMG test showed that muscle nerve function in the Apelin-13 group is better than the HD group. The rotarod test in our investigation shows that better motor recovery is observed in the group having Apelin-13 treatment compared to the HD group. So, Apelin shielded hippocampus neurons from excitotoxicity caused by NMDA receptors [24]. In these neurons, the phosphorylation of Akt on certain residues and Raf /ERK1/2 most likely caused the impact. It was decided that Apelin-13 should be further researched as a possible neuroprotectant against hippocampus damage since. The Apelin/APJ signaling pathway likely serves as an intrinsic survival response for neurons. On the other hand, other studies failed to pinpoint how Apelin-induced myocardial protection involved the PI3K/Akt pathway [25]. It was proposed that Apelin actions might involve other signaling pathways besides Akt/ERK1/2 genes. However, experimental data are still needed to support this hypothesis [26, 27]. Cell counting in immunohistochemistry against the marker Iba-1, a specific marker of microglia cells, shows that treatment with Apelin-13 can reduce microgliosis and as a result, causes a reduction in cell death. There is growing evidence that astrocytes from different neurodegenerative disorders include intracellular aggregates like Syn or HTT. Additionally, these aggregates are known to impair regular astrocytic activity, which has a toxic effect on neurons. IFN and tumor necrosis factor-α (TNF-α), two pro-inflammatory cytokines and chemokines, are crucial in activating astrocytes and microglia [28]. In exchange, it has been demonstrated that activated microglia are a significant biological source of inflammatory and cytotoxic substances, including TNF and interleukin-1 (IL-1), which can cause the death of vascular and neuronal cells. Based on a study, neuroinflammation is implicated in several neurological illnesses [29]. Further, by halting the flow of inflammatory cytokines into the brain, Inhibition of the hepatic NLRP3 inflammasome inhibits DA neuronal degeneration. It appears that inflammation negatively affects neurodegenerative diseases. According to several lines of research [30, 31], Apelin has an anti-inflammatory impact, it has been shown that Apelin-13 treatment reduces activation of NLRP3 inflammasome and the secretion of IL-6, IL-1, and TNF [32]. Furthermore, by reducing inflammation, the loss of APLN hastens the development of increased systolic dysfunction and heart failure [33]. TNF and IL-6 production are inhibited by APJ antagonists [34, 35]. Similar to this, Apelin-13 appears to prevent neurological impairments following an ischemic stroke by reducing inflammation [36, 37]. Apelin’s ability to reduce inflammation is well-established [16]. Immunohistochemically analysis against the astrocytic marker GFAP shows a decreased astrogliosis in the Apelin-13 group. HD is caused by the HTT protein-encoding gene that causes the condition. In addition to increasing the protein’s aberrant function, the mutant HTT also decreases its normal function [38]. Exploring the interplay of the Apelin/APJ molecular pathway in HD, despite the absence of direct evidence impacting HTT proteins, is crucial considering the prodromal molecular events in the condition. Notably, the mutant HTT, a biomarker for HD, serves as an autophagic substrate, and autophagy inducers promote its clearance [39]. Apelin has the potential to expedite the breakdown of HTT via triggering the cellular autophagy process. Moreover, ferroptosis, a process involving lipid metabolism, iron metabolism, and oxidative stress, occurs as a promising healing target for neurodegenerative disease [40]. Apelin-13 can lead to iron accumulation inside the mitochondria thereby triggering ferritinophagy [41]. The Apelin/APJ system, specifically Elabela, can contribute to ferroptosis via influencing the IL-6/STAT3/GPX4 signaling pathway. This process involves scavenging HTT and promoting ferroptosis, suggesting a potential avenue for future treatment strategies in HD [42]. Apelin inhibits the TNF/NF-B pathway, which significantly impacts the delay of HD progression. neuronal death brought on by HTT poisoning in HD fosters inflammation [43]. HD pathogenesis in astrocytes is caused by an increase in the p65-mediated inflammatory response [44]. XPro1595, a human TNF variant lacking TNF receptor-binding activity, has been shown to improve motor function and reduce the quantity of mutant HTT aggregates in R6/2 mice [45]. TNF activates NF-κB. Neurodegeneration brought on by mutant HTT may be caused by abnormal NF-B activation [46]. In medium-sized spiny neurons, the suppression of IkappaB kinase, a crucial NF-B regulator, decreases mutant HTT-induced toxicity [47]. According to growing evidence [48], exogenous and endogenous Apelin exhibit the ability to inhibit the NMDA-induced rise of TNF levels within the retina. Apelin-13 not only hampers the generation of ROS but also possesses the capability to thwart the initiation of the NF-B pathway, as evidenced in both a mouse lung injury [49]. Therefore, it is plausible that Apelin inhibits the growth of HD by obstructing the TNF/NF-B pathway [16].
In conclusion, Apelin-13 exhibits promising therapeutic potential for HD by effectively managing oxidative stress, inflammatory factors, and gliosis processes, while simultaneously enhancing motor function. Considering the interaction of Apelin with the HTT protein in HD, even in the absence of direct evidence, emphasizes the importance of exploring the prodromal molecular events in the disease. Furthermore, Apelin displays anti-inflammatory properties, activation and the secretion of pro-inflammatory cytokines and capability to interrupt the flow of inflammatory cytokines into the brain, aligning with its overall anti-inflammatory impact.
Go to :
 XML Download
XML Download