

 PDF
PDF Citation
Citation Print
Print



Body
Dear Editor,
Autosomal dominant polycystic kidney disease (ADPKD) is the most common monogenic disease leading to end-stage kidney disease [1]. ADPKD mainly arises from genetic alternations in the PKD1 and PKD2 genes, accounting for approximately 75% and 15% of cases, respectively [2]. In comparison to PKD1-related ADPKD, PKD2-related ADPKD presents with milder symptoms and is often diagnosed incidentally through imaging or urologic events at a later age, typically after the fourth decade of life [2].
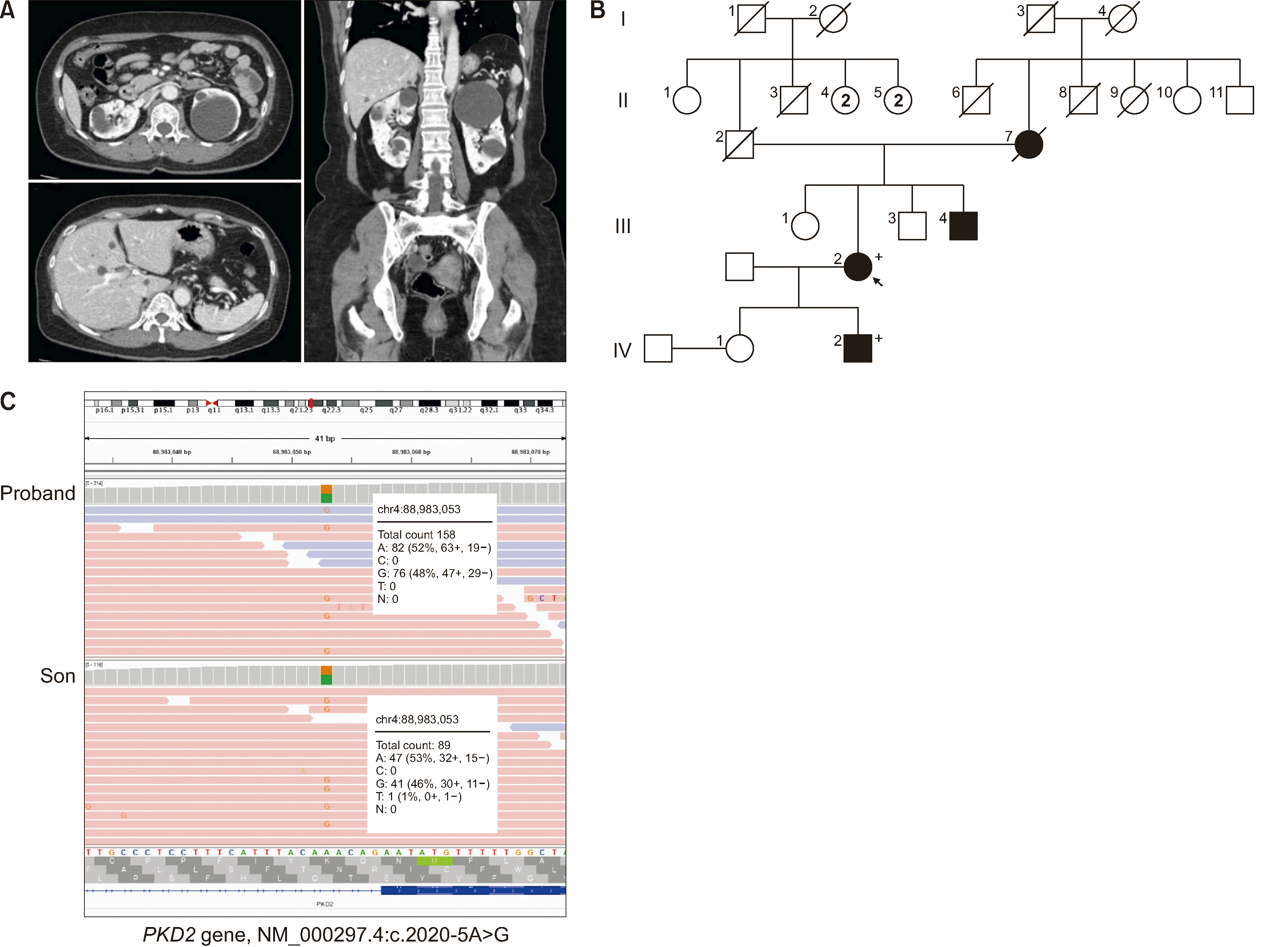
We report the case of a family of patients with ADPKD caused by aberrant splicing in PKD2, from which a 56-yr-old woman presented with multiple renal and hepatic cysts incidentally found on an abdominal ultrasonography performed for a health check-up in April 2019. The institutional review board- of Kyung Hee University Medical Center, Seoul, Korea, approved this study (approval No. 2023-07-078). The patient had a height of 153 cm, weight of 55 kg, and body mass index of 23.5 kg/m2, and had been taking medications for hypertension and hyperlipidemia for 6 yrs. Laboratory investigations showed a normal serum creatinine level (0.82 mg/dL, reference range: 0.34–1.08 mg/dL) and an estimated glomerular filtration rate of 83.4 mL/min/1.73m2. The liver function test was in the normal range, and no specific findings were observed in the urine test. Abdominal computed tomography showed numerous bilateral renal cysts and hepatic cysts of varying sizes (Fig. 1A). The kidneys were enlarged, with a total kidney volume of 563.2 cm3. The mother, brother, and son of the patient had a history of multiple renal and hepatic cysts (Fig. 1B). Her mother also had a history of cerebral hemorrhage (Fig. 1).
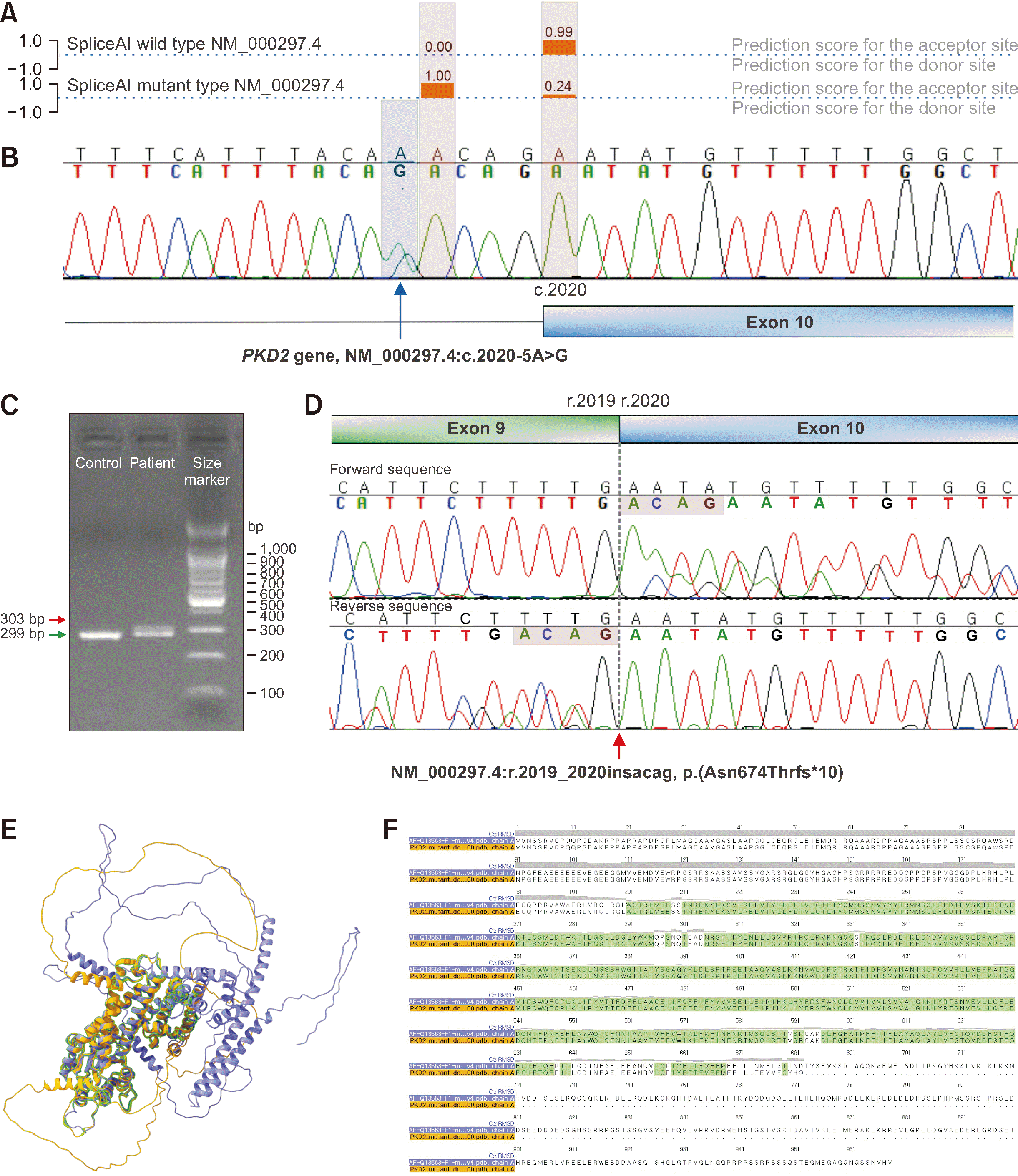
To make a definitive diagnosis of ADPKD, the patient and her son underwent clinical exome sequencing (Fig. 1C). Both individuals had the same heterozygous intronic variant at the acceptor motif, NM_000297.4:c.2020-5A>G, in PKD2. This variant has not been reported in the general population (https://gnomad.broadinstitute.org/) or other patients with ADPKD. Although reported once in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/, last accessed on April 18, 2024), it was classified as uncertain significance (review status: one star) because of insufficient evidence. We predicted the pathogenicity of this variant using SpliceAI [3] and observed a significant change in the acceptor score at position c.2020, decreasing from 0.99 to 0.24 (Delta score: 0.76) (Fig. 2A). In addition, at position c.2020-4, we observed a substantial change in the acceptor score from 0 to 1 (Delta score: 1.00) (Fig. 2A). In summary, this intronic variant is predicted to shift the acceptor site by +1 base pair (bp) instead of causing the typical exon skipping seen with intronic variants near exon boundaries. This shift may potentially induce a premature termination codon (PTC) (Fig. 2).
Therefore, we extracted DNA and RNA from the proband to determine whether there were changes in the RNA sequence as predicted using SpliceAI [3]. We confirmed the presence of the intronic variant using Sanger sequencing (Fig. 2B). Additionally, reverse transcription PCR revealed that, unlike the single band observed in the control (299 bp), our patient had two distinct bands of different sizes (299 bp and 303 bp) (Fig. 2C). Through targeted RNA sequencing, we confirmed that, as predicted using SpliceAI [3], the acceptor site was shifted by +1 bp (NM_000297.4:r.2019_2020insacag), leading to the eventual induction of PTC (p.(Asn674Thrfs*10)) (Fig. 2D) and likely triggering nonsense-mediated decay (NMD) [4].
However, because of the lack of research on NMD efficiency for PKD2, we predicted the structural changes in the mutant PKD2 protein, assuming that the mRNA resulting from this intronic variant might escape NMD and undergo translation. To this end, we compared and analyzed the PKD2 protein structure (yellow) predicted using AlphaFold2 [5] based on the mutant sequence with the canonical PKD2 structure (purple) using ChimeraX Matchmaker (Fig. 2E–2F) [6]. Comparing the canonical and mutant protein structures suggested that some regions would have different structures despite having the same sequence (e.g., residues 1–200). Notably, the region spanning residues 750–785 is crucial for Ca2+ binding (https://www.ebi.ac.uk/interpro/protein/UniProt/Q13563/) and PKD2 channel activity regulation. However, in the mutant protein, this region is not translated, which is predicted to significantly affect protein function [7].
According to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines [8] and ClinGen recommendations (https://clinicalgenome.org/) [9, 10], this intronic variant was classified as likely pathogenic based on the evidence PVS1, PM2_Supporting, and PP1_Supporting. This case, where an intronic variant located outside the ±1 or 2 dinucleotide positions of the donor/acceptor sites induces a PTC owing to intron retention rather than exon skipping [11], highlights the significance of splicing prediction in interpreting intronic variants.
 XML Download
XML Download