

 PDF
PDF Citation
Citation Print
Print



Dear Editor,
Nail-patella syndrome (NPS) is an autosomal dominant (AD) disorder caused by an abnormality in LMX1B and characterized by alterations in the dorsal limb structures, including the nails, knees, elbows, and iliac horns [1]. This report presents the first case of NPS caused by maternal gonosomal mosaicism with a partial deletion of LMX1B. This study was approved by the Institutional Review Board of Seoul National University Hospital Seoul, Korea (H-2204-112-1317).
The proband was a 10-yr-old girl who presented with knee joint pain. She had clinically suspicious nail lesions that appeared at 4 months of age, as well as symptoms characteristic of NPS, including patellar aplasia (Fig. 1), elbow contractures, and iliac horns. In 2013, the patient initially presented with a chief complaint of bilateral elbow joint deformity. Radiographic assessment revealed patellar aplasia; however, the etiology could not be determined at that time. The patient returned in early 2023 and subsequently underwent genetic diagnostic testing. No symptoms suggestive of NPS were identified in the parents, although the brother had characteristics similar to those of the patient. The mode of inheritance observed in this family was inconsistent with that of NPS.
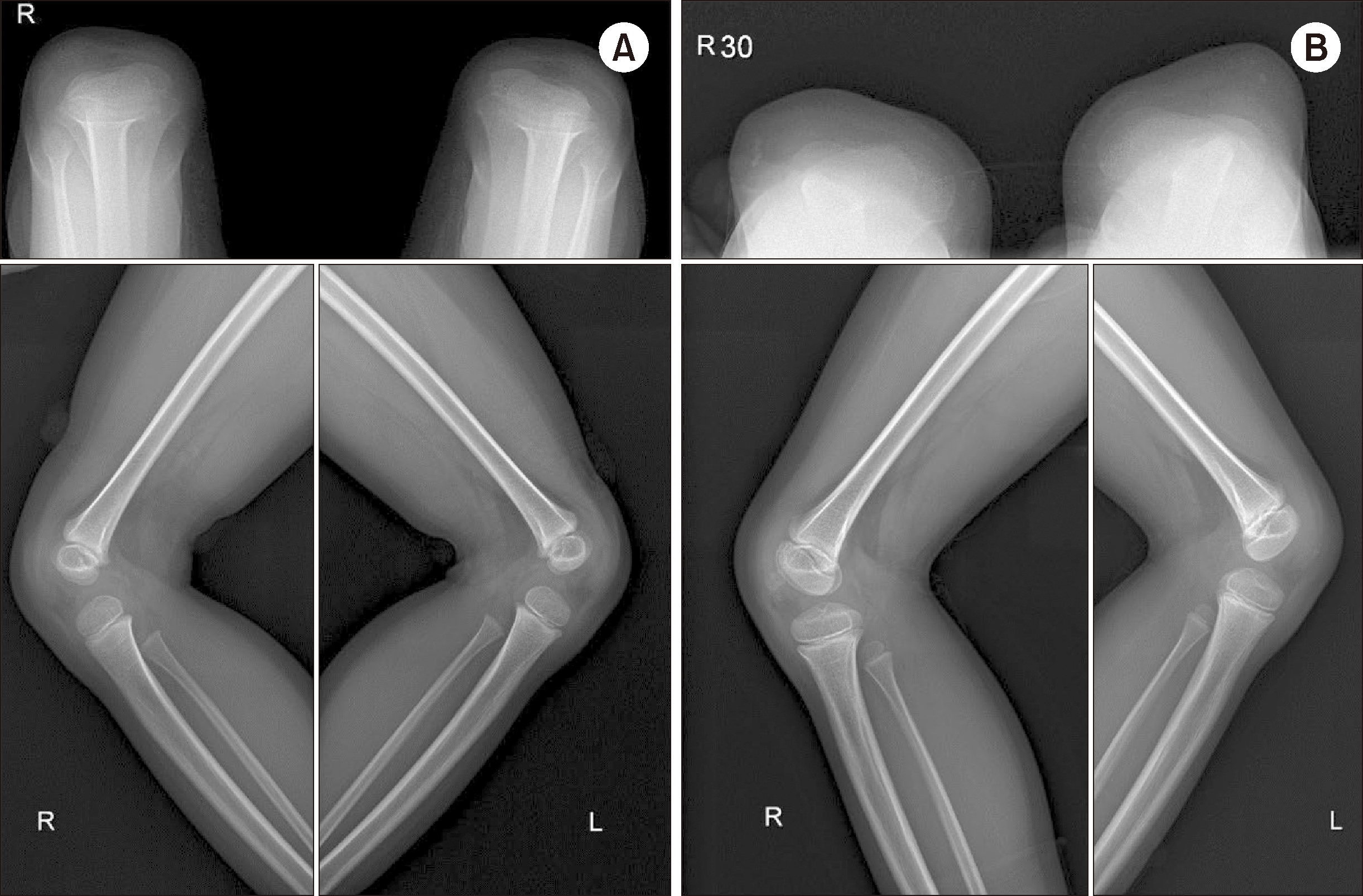 | Fig. 1Plain radiographs of the knees of the proband in skyline (top) and lateral (bottom) views. (A) In 2016, neither patellae was visible. (B) In 2019, limited bilateral patellar ossification without demonstrable normal patella was visible. Images in both panels are suggestive of Nail-patella syndrome.
|
Familial whole-exome sequencing (WES) was performed using peripheral blood samples obtained from the patient, sibling, and parents. WES revealed an approximately 2-kb deletion of LMX1B exons 4–6 in both the patient and her brother. WES data from the patient and her brother revealed that the reads that aligned to exon 4 had soft clips at the same position. The mother’s data revealed the same soft clips in four reads, with a depth of coverage of 299× for the region (Fig. 2A). This finding suggested maternal mosaicism. Familial gap-PCR was performed using primers targeting LMX1B exon 4 and intron 6 to confirm the deletion and primers within the deletion region to confirm the wild-type. The father and a negative control showed identical results. In the patient and her sibling, a band of 648 bp predicted for the deletion and a normal 489-bp band were observed, confirming a heterozygous deletion. In the patient’s mother, both bands were observed, although the 648-bp band was dim, confirming a mosaic deletion. Sanger sequencing helped identify the same breakpoint as determined using WES (Fig. 2B–D). The variant was NM_001174147.2(LMX1B):c.641_887-556delinsGG, p.(Lys214Argfs*11). According to the 2015 American College of Medical Genetics and Genomics (ACMG) guidelines [2] and ACMG/ClinGen technical standards [3], it was classified as pathogenic.
 | Fig. 2Integrative Genomics Viewer snapshot of the family (A) and gap-PCR design and results (B–D). (A) Clockwise from top left: the proband (GL00285P), father (GL00285F), mother (GL00285M), and brother (GL00285S). The proband and her brother have right-clipped reads (red box) harboring the same breakpoint partially aligned to exon 4 of LMX1B. This right-clipped read is also observed for the proband’s mother (red box) but not the father. (B) Three 20-mer oligos were used to design a PCR assay for the detection of a deletion. EX-4F (purple arrow) is a common forward primer, and EX-4R and IVS-6R (red arrows) are reverse primers for wild-type detection and deletion confirmation, respectively. LMX1B consists of eight exons, with a DNA-binding domain spanning exons 4–6. Loss of this domain was predicted in this family. (C) A band of 648 bp, the amplicon of the EX4-F/IVS6-R primer pair, and a band of 489 bp, the product of the EX4-F/R primer pair, were identified in the proband (GL00285P), her brother (GL00285S), and their mother (GL00285M). Therefore, a deletion spanning exons 4–6 was confirmed. In the father (GL00285F), a 648-bp band was not observed, indicating no deletion. The negative control (NC) also did not have a 648-bp band. (D) The 648-bp PCR product was extracted and Sanger-sequenced. A breakpoint was identified between c.640 and c.887-555 in the electropherogram.
|
To the best of our knowledge, this is the first report of NPS caused by an inherited gonosomal mosaic deletion of LMX1B in unaffected parents. No pathogenic variants other than this deletion were identified, confirming that the deletion was the cause of NPS. To determine the cause of the deletion, we analyzed the surrounding sequence and identified a homologous sequence flanking the breakpoint, which consisted of ATGAC(N)4-5GG. Therefore, we inferred that a double-strand break had occurred, followed by microhomology-mediated end joining [4]. LMX1B is a member of the LIM-homeobox domain (HD) family, which is responsible for protein–protein interactions and DNA binding. Loss of the HD, encoded by exons 4–6, causes it to function as a transcription factor. A case of NPS caused by a deletion within the HD has been reported, supporting this theory [5], and an experimental study has demonstrated that mutations within the HD cause the loss of DNA binding and reduced transcriptional activity [6].
Autosomal recessive genetic disorders are typically associated with loss of function, whereas dominant genetic disorders are associated with dominant-negative (DN) effects or gain of function (GOF) [7]. However, haploinsufficiency in AD genetic disorders has a more disruptive effect on protein structure than GOF/DN [7]. NPS follows an AD inheritance pattern, but its molecular pathogenesis shows haploinsufficiency [8]. We identified identical deletions in the siblings, suggesting gonosomal mosaicism. The probability of a de novo variant being shared by siblings is approximately 1.1%; however, in the presence of mosaicism, it increases to approximately 23% [9]. The risk of transmission of maternal mosaicism reportedly is higher than that of paternal mosaicism [10]. The risk of recurrent transmission is stable regardless of the maternal age but decreases with the paternal age [10]. Individuals with an NPS-causative heterozygous LMX1B variant exhibit high penetrance [1]. NPS is managed according to the unique manifestations in each patient. Therefore, genetic counseling should be offered to patients and mosaic parents. This study was limited by the lack of testing of saliva or fibroblasts. However, in a previous study, the rate of mosaicism in the blood was higher than that in other tissues, including hair and saliva [1].
In conclusion, we report a case of familial NPS caused by a heterozygous deletion of exons 4–6 of LMX1B inherited from an asymptomatic carrier mother with gonosomal mosaicism of LMX1B. This indicates that gonosomal mosaicism may significantly contribute to disease transmission, even in asymptomatic parents. Consequently, developing more advanced genetic counseling techniques that consider this factor is imperative to improving the management and prevention of genetic diseases, such as NPS, in families with potential gonosomal mosaicism.
 XML Download
XML Download