

 PDF
PDF Citation
Citation Print
Print



Introduction
Pregnancy is a natural process that can, unfortunately, lead to traumatic events. Various defects or alterations in the normal pregnancy process can result in miscarriages or abortions (spontaneous or threatened). Miscarriage is defined as the spontaneous demise of pregnancy before the fetus reaches viability. This includes all cases of pregnancy loss from the time of conception to 24 weeks of gestation [1]. Recurrent miscarriage (RM) can be defined as two or more successive spontaneous losses of pregnancy, affecting 5% of women in the early stages of pregnancy. However, nearly 1% of women experience three or more consecutive pregnancy losses, including second-trimester abortions [2].
RM is categorized into three types: 1) primary RM or first-trimester loss; 2) secondary RM or mid-trimester loss; and 3) tertiary RM or late fetal loss. RM is considered multifactorial due to its diverse causes, including anatomical factors (vascular interruption of the endometrium, intrauterine adhesions, uterine fibroids, polyps, inadequate placentation, and congenital uterine abnormalities), endocrine factors (diabetes mellitus, hypothyroidism, hyperprolactinemia, luteal phase defect, polycystic ovarian syndrome, and inadequate production of progesterone by the corpus luteum), as well as cytogenetic and uterine anomalies. Other contributing factors include immunological conditions (antiphospholipid antibody syndrome [APS], presence of lupus anticoagulant [LAC], and anti-cardiolipin antibody [ACA]), infections, untreated hypothyroidism, high sperm DNA fragmentation levels in males, stress, and lifestyle choices [3,4]. RM is also associated with socio-psychological trauma and financial burdens on society. Unfortunately, approximately 50% of RM cases remain unexplained due to unexplored markers responsible for RM. To reduce the incidence of pregnancy loss, various screening tests are suggested for early and accurate diagnosis. These tests include: antiphospholipid antibody test (immunologic), glucose tolerance test (metabolic), thyroid stimulating hormone test (endocrine), ultrasonography (clinical), chromosomal assessment (genetic), and tests related to vascular etiology [5,6].
Thrombophilia is an abnormality in the cascade of proteins responsible for blood coagulation, increasing the risk of thrombosis (blood clots in blood vessels). The predominant manifestations related to thrombophilia are pulmonary embolism and deep vein thrombosis, collectively known as venous thromboembolism [7]. Thrombophilia plays a major role in RMs, primarily during the onset of pregnancy. An ideal balance between coagulation and fibrinolytic factors is essential for a successful pregnancy. This balance prevents excess fibrin aggregation and clot formation within the placental blood vessels, and is crucial for proper fibrin polymerization and stabilization of the placental basal plate during embryo implantation. Defective pregnancy outcomes are typically related with maternal thrombophilia but can sometimes also be associated with fetal thrombophilia caused by the inheritance of paternal or maternal thrombophilic genes [8]. Thrombophilia is classified into two types: inherited and acquired. Inherited thrombophilia is an intrinsic condition present at birth that predisposes the individual to develop thrombosis, whereas acquired thrombophilia develops later in life. The present review addresses various thrombotic factors and their accepted etiologies, genetic interactions, prognosis, possible therapeutics, and prospects for RM. It reveals the genetic landscape of thrombophilia in RM based on different approaches, including the candidate gene studies, meta-analyses, genome-wide association studies (GWAS), and other high-throughput methods. Understanding the contribution of both inherited and acquired mutations to pregnancy mechanisms would further improve our understanding of RM and aid in the development of targeted therapy-based molecular treatments.
Thrombotic etiologies
Thrombotic events causing thrombophilia can interfere with the transfer of essential nutrients from maternal blood to fetal tissues, leading to RM. Factors associated with heritable thrombophilia include factor V Leiden (FVL) polymorphism, protein C and S deficiencies, and antithrombin (AT) and prothrombin mutations. On the other hand, acquired thrombophilia associated with RM includes conditions, such as hyperhomocystenemia and resistance to activated protein C resistance (APCR) [9,10].
Inherited thrombophilia
Inherited thrombophilia refers to conditions that increase the risk of thromboembolism due to hereditary changes in functional proteins involved in coagulation. The most common causes of inherited thrombophilia include: FVL and prothrombin G20210A mutations, methylene tetrahydrofolate reductase (MTHFR) deficiency, AT deficiency, and protein C and S deficiencies. Although these mutations and deficiencies are linked to an increased risk, it is important to note that women with homozygous FVL or prothrombin mutations generally have a two-fold risk for early pregnancy complications, such as intermittent premature delivery in the first trimester [11].
Mechanism of thrombophilia
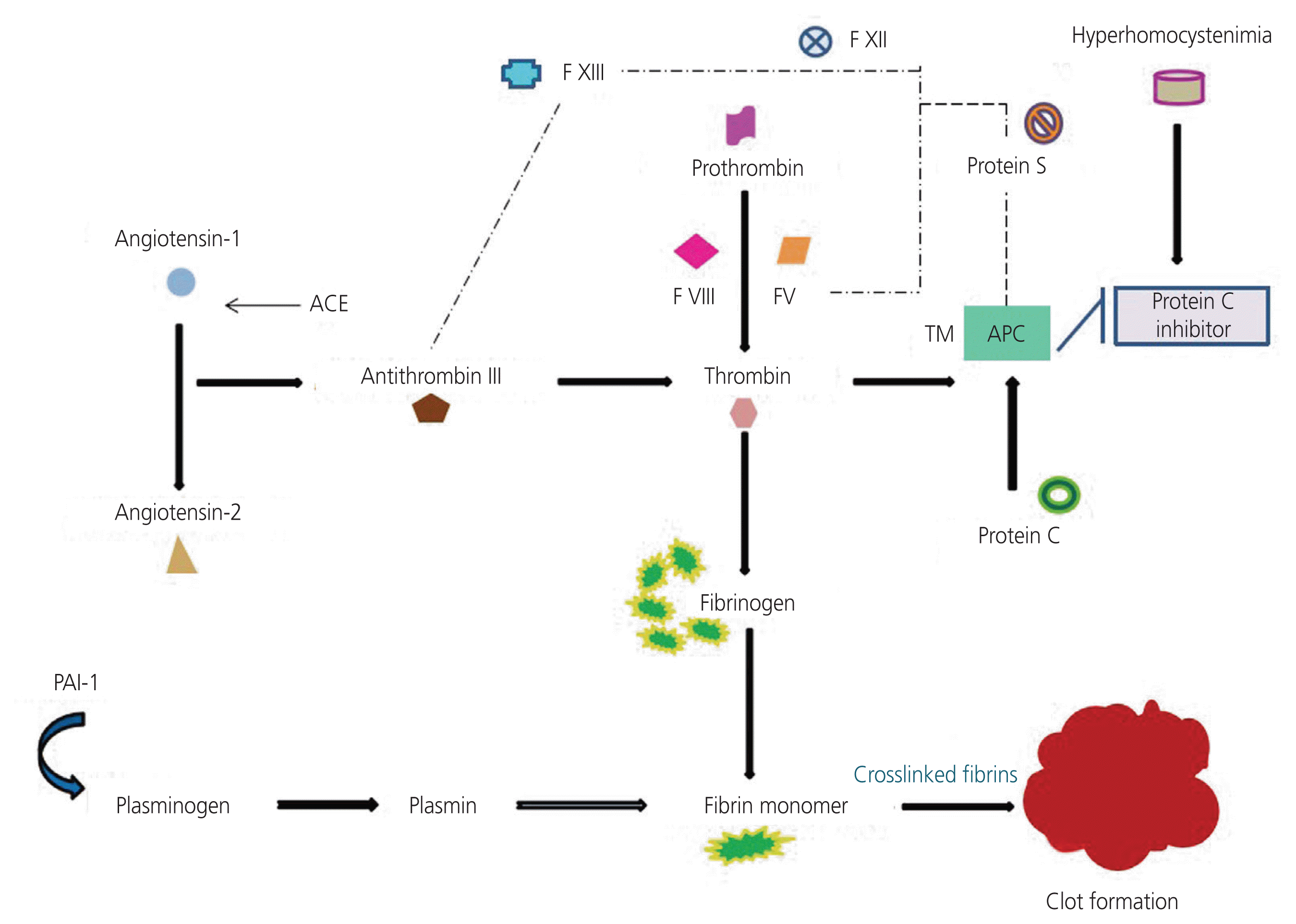
Thrombophilia arises from either the failure to inactivate thrombin or the inability to control thrombin protein formation. AT binds to heparin sulfate or endothelial cells, deactivating thrombin, factor XIa, factor IXa, and factor Xa, thereby maintaining blood fluidity. Protein C, an anticoagulant, regulates thrombin generation and is activated when thrombin binds to thrombomodulin in the veins [12]. Other thrombophilic components associated with RM include angiotensinogen, fibrinogen, and AT. FVL (factor V-1q23) represents the most widely recognized polymorphism associated with RM. FVL may cause thrombosis by reducing factor V’s capacity to deactivate activated protein C (APC), leading to increased thrombin production. APCR prolongs thrombin degradation, potentially indicating a hypercoagulable state. Decreased AT activity hampers the thrombin inactivation and reduces the efficiency of protein C or protein S, further limiting the control of thrombin formation. These mechanisms heighten susceptibility to venous thrombosis and RM [13,14]. The beta-fibrinogen (455 G/A) polymorphism is linked to elevated plasma fibrinogen levels and may enhance the enzyme substrate interaction involving thrombin, fibrinogen, and platelets [15]. This could lead to increased intravascular fibrin coagulation, raising the risk of placental thrombosis that may trigger RM [16]. Fig. 1 illustrates the coagulation pathway involving both pro- and anticoagulant factors.
Acquired thrombophilia
Acquired thrombophilia represent hypercoagulable states associated with an increased risk of thromboembolism and miscarriage. APS is the predominant type of acquired thrombophilia and is associated with both venous and arterial thrombosis [17]. Recent findings indicate that acquired thrombophilia is more common in women with RMs, while inherited thrombophilia, caused by FVL and prothrombin G20210A mutations, is less common in the Indian population [18].
APS
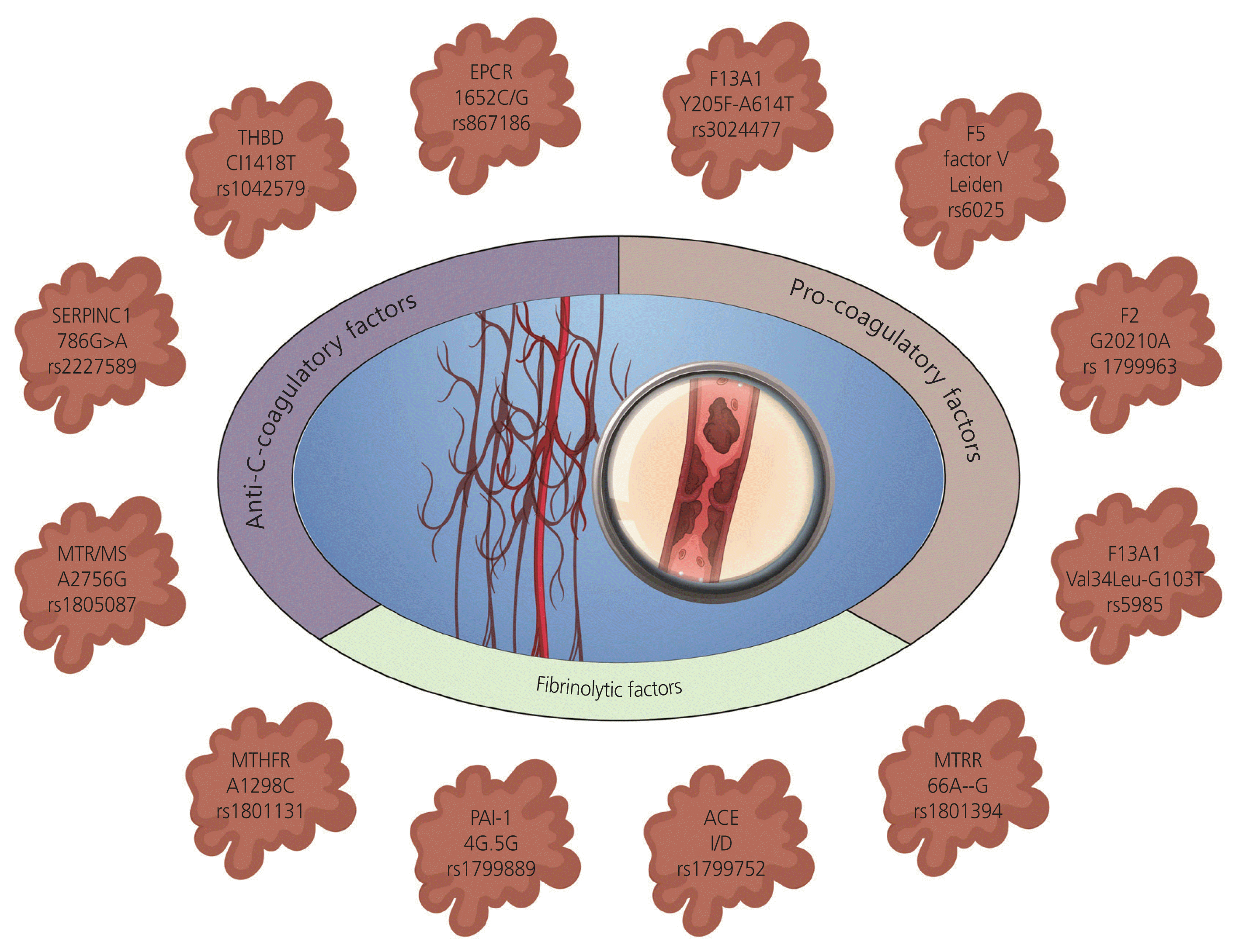
APS is a type of thrombophilia that develops later in life and is not inherited. In APS, the body produces antibodies against phospholipid-bound proteins that harm vessels by forming blood clots, leading to venous thrombosis, preterm delivery, placental inadequacy, preeclampsia, and miscarriages. APCR is the second most prevalent form of acquired thrombophilia, affecting 5–20% of women with RM. Sedano-Balbás et al. [19] have demonstrated a relatively high prevalence of antiphospholipid antibodies in individuals from the general population suffering from vascular occlusions or pregnancy complications. Both inherited and acquired thrombophilia are associated with an increased risk of miscarriage. The main genes involved in the fibrinolytic, procoagulant, and anticoagulant pathways responsible for RMs are shown in Fig. 2.
Coagulation
Factor V plays an important role in maintaining hemostasis through both the procoagulant and anticoagulant pathways. Prothrombin is converted into the serine protease thrombin by the prothrombin convertase complex, which includes activated factors V and X. Thrombin then initiates three major steps in fibrin development: 1) building fibrin monomers; 2) accumulating these monomers; and 3) crosslinking fibrin glutamine residues to form an insoluble clot, a process that requires transglutaminase factor XIII. A key mechanism of feedback inhibition for prothrombin convertase involves the combined action of APC and protein S bound to phospholipid surfaces. This leads to the degradation and inactivation of factors V and VIII [19].
Procoagulant factors
1. Factor V
Dysfunctional factor V can greatly affect hemostasis. APCR is caused by a point mutation in the factor V gene, manifested in a heterozygous state in 5–10% of individuals. Factor V has been demonstrated to exhibit two point mutations: H1299R and Y1702C. The H1299R mutation is associated with low APCR and presents an additional risk for FVL carriers, whereas the Y1702C mutation leads to factor V deficiency. Maternal thrombophilia caused by FVL polymorphism may induce microthrombosis in placental blood vessels, resulting in placental infarctions. These infarctions can damage the maternal blood vessels that supply the placenta, leading to low placental perfusion and eventually fetal death [20]. A meta-analysis by Eslami et al. [21] reported an association between the FVL 1691G >A mutation and RM, including a subgroup analysis based on continents. The results showed a substantially increased risk of RM in Asian, African, and European populations, but not in South American populations [21].
2. Factor II (prothrombin)
The G20210A mutation in the prothrombin gene leads to a coagulation disorder. Factor II converts fibrinogen into fibrin to form thrombi, stimulates platelet development, and activates factors V, VIII, XIII, and protein C, thereby regulating coagulation. The prothrombin dimorphism G20210A is the second most common type of inherited thrombophilia that leads to increased concentrations of prothrombin in the blood. A study reported that elevated prothrombin levels may lead to abnormalities in placental function, affecting cell attachment, smooth muscle cell proliferation, and vasculogenesis [22].
3. Fibrinogen
Fibrinogen is a glycoproteins involved in blood clotting. The morphology of blood clots, including their structure, size, and stability, is strongly influenced by conditions present during fibrin generation. These conditions include the levels of procoagulants, anticoagulants, fibrinogen-binding proteins, and metal ions. Variations in fibrinogen molecule’s three associated polypeptide chains (α, β, and γ) can lead to changes in clot properties. Polymorphism in the β-chain of fibrinogen (−455 G/A) has been independently associated with plasma fibrinogen levels. This polymorphism improves enzyme-substrate interactions between coagulatory particles, resulting in expanded intravascular fibrin deposition, leading to placental thrombosis and RM [23].
4. Factor XIII
Transglutaminase factor XIII is activated by thrombin and stabilizes fibrin clots by covalently crosslinking fibrin monomers. The factor XIII and Val34Leu polymorphism has been associated with thrombosis. Patients with thrombosis frequently exhibit Val/Val genotype, while the Val/Leu phenotype is less common. The Leu34/Leu genotype provides a strong defense against thrombosis [24]. Individuals homozygous for factor XIII are also known to have a high risk of recurrent premature birth. Factor XIII deficiency leads to delayed wound healing, severe bleeding, and a high risk of premature delivery in women.
Anticoagulatory factors
1. Protein C and protein S
APC degrades activated factors V and VIII, making it a crucial component of the anticoagulatory mechanism. APC formation occurs when protein C is activated by the thrombomodulin-thrombin complex. Protein S serves as a cofactor for APC, enhancing its ability to restrain blood clotting. Deficiency in protein S increases the risk of thrombosis [25]. Protein C deficiency involves more than 160 different mutations in the gene encoding protein C located on chromosome 2 (2q13–14) and is inherited as a autosomal dominant defect. Protein S enhances the effects of protein C on factors V and VIII, thereby suppressing thrombin formation [26,27]. Mekaj et al. [28] reported that the prevalence of protein S deficiency in healthy individuals is 0.03–1.3%, with the associated risks similar to those of protein C deficiency. The incidence of AT, protein C, and protein S deficiencies, as well as FVL, is higher in patients with thromboembolic events than in the healthy population [28].
2. AT III
AT III is a plasma serine protease inhibitor that hinders thrombin activity by covalently binding to its proteolytic site, thereby maintaining blood fluidity. Congenital AT III deficiency is an autosomal dominant disorder, wherein an individual inherits a copy of the gene encoding AT III on chromosome 1q25.1 [27]. AT is an important physiological regulator of fibrin development and inhibits thrombin. The mutation in the thrombin encoding gene is inherited as an autosomal dominant trait. The mutated protein directly binds to factors XI and X, as well as thrombin, thereby reducing the level and activity of AT. The mutation also alters the structure and function of AT, diminishing its activity [29,30].
Plasminogen activator inhibitor-1 (PAI-1)
PAI-1 is present in plasma, platelets, endothelial cells, vascular smooth muscle cells, and the extracellular matrix. PAI-1 is the primary physiological inhibitor of tissue-type plasminogen activator and urinary-type plasminogen activator, both key regulators of fibrinolysis. A single nucleotide deletion/ insertion occurs 675 nucleotides upstream of the PAI-1 transcription start site, producing two alleles: PAI-1 (−675 4G) and PAI-1 (−675 5G), which brings about unequal fibrin accumulation [31]. Hypofibrinolysis, caused by the PAI-1 4G/5G polymorphism, is a critical factor for pregnancy complications, including preeclampsia, intrauterine growth restriction, and stillbirth. PAI-1, a crucial inhibitor of fibrinolysis, may reduce plasmin-dependent proteolysis, inducing repeated abortion by stimulating fibrin during early placental development [32,33].
Angiotensin converting enzyme (ACE)
Hereditary control of plasma ACE levels can be examined by detecting the Ile/Asp polymorphism that results in variable ACE levels. The renin-angiotensin system maintains the proper balance between vasoconstriction and vasodilation. ACE converts angiotensin I to angiotensin II. Angiotensin II has vasoconstrictor properties and regulates vascular tone [34]. Variations in angiotensin II concentration may affect the placental circulation regulation and are considered as risk factors for RM [35].
Factor XII
Coagulation factor XII, also known as the Hageman factor, is a single-chain zymogen without any evident enzymatic activity. Patients with factor XII deficiency appear to develop thromboembolic diseases. The C46T polymorphism in the 5′ untranslated region of the gene encoding factor XII affects translation regulation, leading to lower plasma levels. Factor XII deficiency plays a role in gestation, delivery, and RMs [36].
MTHFR
MTHFR, located on chromosome 1p36.3, catalyzes the remethylation of homocysteine to methionine. Elevated homocysteine levels are observed in individuals with RM who are homozygous for the MTHFR C677T mutation, especially in the presence of folate deficiency [37]. Hyperhomocysteinemia is a risk factor of cerebrovascular, vascular, and coronary diseases. The impact of the C677T mutation in MTHFR on RMs or pregnancy-related issues is controversial. Patients with RM before 17 weeks of gestation are several times more likely to be homozygous for MTHFR than controls [38]. Parveen et al. [37] reported major susceptibility effects of MTHFR C677T, A1298C, and MTHFD G1958A mutations among Asian patients with RM.
Homocysteine
Homocysteine, produced from the metabolism of the amino acid methionine, circulates in the plasma at concentrations of 5–16 mol/L. Various factors are associated with hyperhomocysteinemia, including severe preeclampsia, stillbirths, and RMs [39]. Deficiencies in folate, vitamins B6 and B12, or inherited enzymatic defects, can also result in hyperhomocysteinemia [40].
Genetics of inflammation and thrombophilia in RM
Immune cells and inflammatory molecules play central role in thrombosis. The relationship between inflammation and plasma coagulation is an intricate network that balances an organism’s response to injury and pathogen invasion while maintaining homeostasis. Dysregulation of this balance can lead to pathological thrombosis and organ damage. The pathophysiological link between inflammation, the fibrinolytic system, and miscarriage involves a systemic and local imbalance of inflammatory mediators, inflammatory infiltration of the endometrium, fibrosis, thrombosis, and endothelial dysfunction, resulting in poor vascularization following implantation. Significant associations have been reported between RM and polymorphisms in platelet integrins, increased platelet aggregation, and thrombus formation in the intervillous space [41,42]. A large body of evidence suggests that endothelial dysfunction, a condition of low-grade inflammation, is one of the earliest indicators of thrombotic phenomena. It has been reported that the anti-inflammatory Th2 cytokines (e.g., interleukin-10) can exert a protective role in pregnancy, whereas the pro-inflammatory Th1 cytokines (e.g., interferon-γ and tumor necrosis factor-α) have deleterious effects on pregnancy outcomes, including fertilization, implantation failure and miscarriages [43].
Genetic association with RMs
Most of the available literature has evaluated the role of alterations in several genes associated with thrombophilic pathway, inflammatory pathway, DNA repair pathway, metabolic pathway, and microRNAs in the context of RM. However, the small sample size of these studies has been a major limitation. Nevertheless, multidimensional reports and meta-analyses have somewhat overcome this limitation (Table 1).
Gene-gene interaction
Candidate gene studies have revealed moderate associations with RM, primarily due to variations in RM definitions and participant selection criteria, necessitating consensus. Coagulation and anticoagulation genes are natural candidates for RM susceptibility. Numerous studies have suggested that FVL and prothrombin 20210A mutations are frequently implicated in RM, with compound heterozygotes for AT, protein C, and protein S being exceptionally rare. One study reported a mild risk of thrombosis in patients with either heterozygous FVL or prothrombin mutations, with risks approximately 3.8 and 4.9 times higher, respectively, for blood clots. Carriers of both heterozygous mutations may face a 20-fold increased risk [44]. FVL mutation alone accounts for 20–25% of isolated thrombotic events and 40–45% of inherited thrombophilia cases associated with fetal loss. A deletion/insertion polymorphism (4G/5G) in SERPINE1 affects its transcriptional regulation, altering gene expression dynamics [45]. Although these single nucleotide polymorphism (SNP)-based studies can only provide an indication of association, their clinical utility in decision-making remains limited. Thus, large-scale replication studies and GWAS are recommended.
Multi-gene (pathway) studies
There is limited literature available on the influence of gene-gene interactions on the risk of RM. Studies using multidimensional reduction and classification and regression tree analysis have explored these interactions. However, a study focusing on the combined effect of genetic variants in thrombophilic, inflammatory, obesity, and detoxification pathways highlighted the importance of interleukin-10 and leptin gene polymorphisms in predisposing individuals to RM [46]. Several comparative studies have consistently identified several clinically relevant polymorphisms in patients with RM, including thrombophilia-associated FVL mutation, prothrombin G20210A mutation, and the MTHFR C667T variant affecting enzyme activity [47].
GWAS-based approach
Most initial association studies in RM focused on candidate genes, examining one or a few genetic loci with disease risk. However, due to limited validation studies, these approaches did not yield as robust outcomes as anticipated by epidemiologists and researchers. GWAS have since emerged as powerful tools for scanning genetic variants across the entire genome in diverse populations. They enable association studies using sets of SNPs that tag common variants without prior knowledge of their function or location. Numerous GWAS have been conducted on RMs, suggesting that a broad array of thrombophilic and inflammation-related genetic variants, rather than SNPs, collectively contribute to the risk of RM. For instance, inter-alpha-trypsin inhibitor heavy chain 4, a positive regulator of interleukin-6 involved in the implantation process, shows reduced expression levels in patients with RM [48,49]. Recently, Laisk et al. [50] integrated GWAS results from up to 69,054 cases from different ancestries (European, Chinese, UK South Asian, UK African, African American, Hispanic American, and UK Caribbean) for sporadic and multiple consecutive miscarriages to study maternal genetic susceptibility and underlying biological mechanism of miscarriage. These findings suggest that genetic variations potentially related to placental biology contribute to the etiopathogenesis of miscarriages [50].
High-throughput approaches
RM is often associated with multiple genetic mutations, necessitating the identification of driver mutations that lead to its pathogenesis. The advent of GWAS and high-throughput sequencing methods, such as next-generation sequencing (NGS), has remarkably advanced our understanding of the genetic and mutational landscape of RMs [51]. However, GWAS has some limitations: the data are cumbersome and expensive, and the majority of scanned SNPs are located in the intronic regions of the genome, complicating functional validation. Additionally, GWAS often fail to detect rare variants, such as those causing deficiencies in protein C, AT, and protein S [52]. NGS, particularly whole-genome sequencing (WES), has provided a platform for identifying genes responsible for miscarriages by searching for genetic changes at the nucleotide level within the entire genome or in the coding part of the genome (exome). Unlike GWAS, high-throughput sequencing methods, especially WES, are cheap and easy to process, and the scanned SNPs are located in the exonic regions of the genome [53]. However, few studies have specifically explored the genetic landscape of RM using NGS. One study identified a heterozygous mutation in KIF14, which causes miscarriage, by performing WES on a trio (a couple and one of the recurrent losses exhibiting microcephaly, renal, skeletal, and craniofacial abnormalities) [53]. NGS and bioinformatics analyses identified four mutations in three genes involved in coagulation pathways: coagulation factor V-F5 c.4619A>C (p.Glu1512Ala), F5 c.5932A>C (p.Thr1950Pro), thrombomodulin-THBD c.457T>G (p.Trp153Gly), and fibrinogen alpha chain FGA c.2054T>G (p.Phe685Cys) [54].
Research gaps
RM presets a significant challenge due to its high incidence across various geographic regions. To effectively address this complex issue, it is crucial to comprehensively understand the genomic landscape of RM, complemented by transcriptomics and proteomics data. These insights can pave the way for preclinical trials aimed at developing targeted therapeutics. Currently, therapeutic options for RM are limited, highlighting the need to leverage driver mutations for targeted therapy approaches that could improve clinical outcomes. Despite these efforts, there are no clinically approved drugs specifically designed for RM treatment at present [55].
Diagnosis in context to RM
Diagnosing RM requires meeting specific clinical and laboratory criteria. These criteria include one or more confirmed episodes of vascular thrombosis (venous, arterial, or small vessel) and pregnancy complications, such as preeclampsia, placental insufficiency, and RM.
Laboratory evaluation
Testing must be positive on at least two assessments, taken 1 or 6 months apart. The recommended laboratory tests are as follows. 1) The recommended tests for thrombophilia screening include, determination of prothrombin time, reptilase time, LAC fragmented thromboplastin time, thrombin time, anti-β2 glycoprotein 1 antibody levels, APCR, FVL and prothrombin mutations, fibrinogen levels, and basal homocysteine levels. 2) Vascular thrombosis is diagnosed by at least one clinical episode of blood vessel (venous or arterial) thrombosis confirmed by imaging or histopathology. 3) For hereditary examination of pregnancy-related tissues, exhibit-based near genomic hybridization is recommended [56]. 4) Screening for antiphospholipid antibodies, LAC, and ACA (immunoglobin G and immunoglobin M) should be performed after two miscarriages [57]. 5) Abnormal thyroid-stimulating hormone and thyroid peroxidase levels should be followed up with thyroxine testing in women with RM. 6) Sonohysterography is more accurate than hysterosalpingography for diagnosing uterine deformities and is used to assess uterine morphology when 3-dimensional ultrasound is not accessible [58]. And 7) hyperhomocysteinemia should be analyzed by estimating fasting homocysteine levels using gas chromatography-mass spectrometry or other biochemical methods.
Treatment
1. Treatment for inherited thrombophilia
Aspirin or heparin treatment improves pregnancy outcomes in women with RM and inherited thrombophilia. Pregnant patients with AT deficiency, or those who are homozygous or heterozygous for FVL or prothrombin (G20210A) mutations, require therapeutic unfractionated or low-molecular-weight heparin treatment throughout the fetal developmental period [59]. Patients with low thrombogenic thrombophilia should be receive prophylactic heparin antepartum. For hyperhomocysteinemia, the focus should be supplementation with folic acid and vitamins B6 and B12 throughout pregnancy. However, antithrombotic prophylaxis is uncommon in hereditary thrombophilia [60,61].
2. Thrombophilia and RM
Antithrombotic prophylaxis is not recommended in women with RM and inherited thrombophilia. However, for women with RM and APS, low-dose aspirin (75–100 mg/day) before conception and prophylactic heparin treatment are proposed after a positive pregnancy test [62]. Women with RM suspected of having an immunological history should receive high-titer antiphospholipid antibody treatment to increase the likelihood of live births [63].
Conclusion
Managing thrombophilia during pregnancy is complex and requires a delicate balance between the coagulation pathways. Anticoagulation therapy may benefit women both as a prophylactic measure and as a treatment for RM. The prognostic value of various novel biomarkers must be evaluated by integrating genomic, proteomic, and metabolomic data with clinical information into a multivariate analysis model. Therefore, the effectiveness of these biomarkers needs to be validated in large clinical trials. The prevalence of thrombophilia in women with RM is higher than in the general population. This study demonstrated that preventive treatment using high-throughput technologies and medications, such as enoxaparin, aspirin, heparin, and folic acid, can improve the rate of live births in women with or without thrombophilia.
 XML Download
XML Download