

 PDF
PDF Citation
Citation Print
Print



Introduction
Heavy-chain deposition disease (HCDD) is a rare, nonorganized monoclonal immunoglobulin (Ig) deposition disease [1]. From 1992 to 2020, around 70 cases of HCDD were documented [2]. In a retrospective observational study at the Mayo Clinic spanning from 1992 to 2011, seven cases of HCDD were diagnosed, representing 0.07% of all renal biopsies [3]. Unlike common light-chain Ig deposition diseases, HCDD infrequently co-occurs with multiple myeloma (MM) [4]. Due to the rarity of the disease and the challenges associated with its diagnosis, the prognosis is generally poor. Diagnosis is often delayed; in one study, it took an average of 1 year from the onset of the disease to reach a diagnosis [5]. Therefore, to improve the prognosis of the kidney through prompt diagnosis and treatment, we report a rare case of HCDD with MM in which the patient showed rapidly progressive renal failure, nephrotic-range proteinuria, and hematuria.
Case
Ethical statements: Our institution does not require Institutional Review Board approval for case reports. Informed consent was obtained from the patient for the publication of this case report and accompanying images.
A 56-year-old businessman with a history of hypertension, heart failure, and coronary artery disease presented at an outpatient clinic with complaints of whole-body pain, edema, dizziness, dyspnea, and blood-tinged sputum that had persisted for 2 weeks. A physical examination revealed anasarca. He had been using nonsteroidal anti-inflammatory drugs for the past 2 weeks to manage headaches.
In the initial assessment, the following results were observed: a hemoglobin level of 8.2 g/dL, a blood urea nitrogen/creatinine ratio of 84.8/3.14 mg/dL, a CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) estimated glomerular filtration rate (eGFR) of 21 mL/min/1.73 m2, and levels of total protein, albumin, and beta-2-microglobulin at 20.9 mg/L, 3.1 g/dL, and 20.9 mg/L, respectively. Urinalysis revealed a random urine protein/creatinine ratio of 3,527 mg/g, a random urine albumin/creatinine ratio of 3,070 mg/g, 21–30 red blood cells per high-power field, and 11% dysmorphic red blood cells.
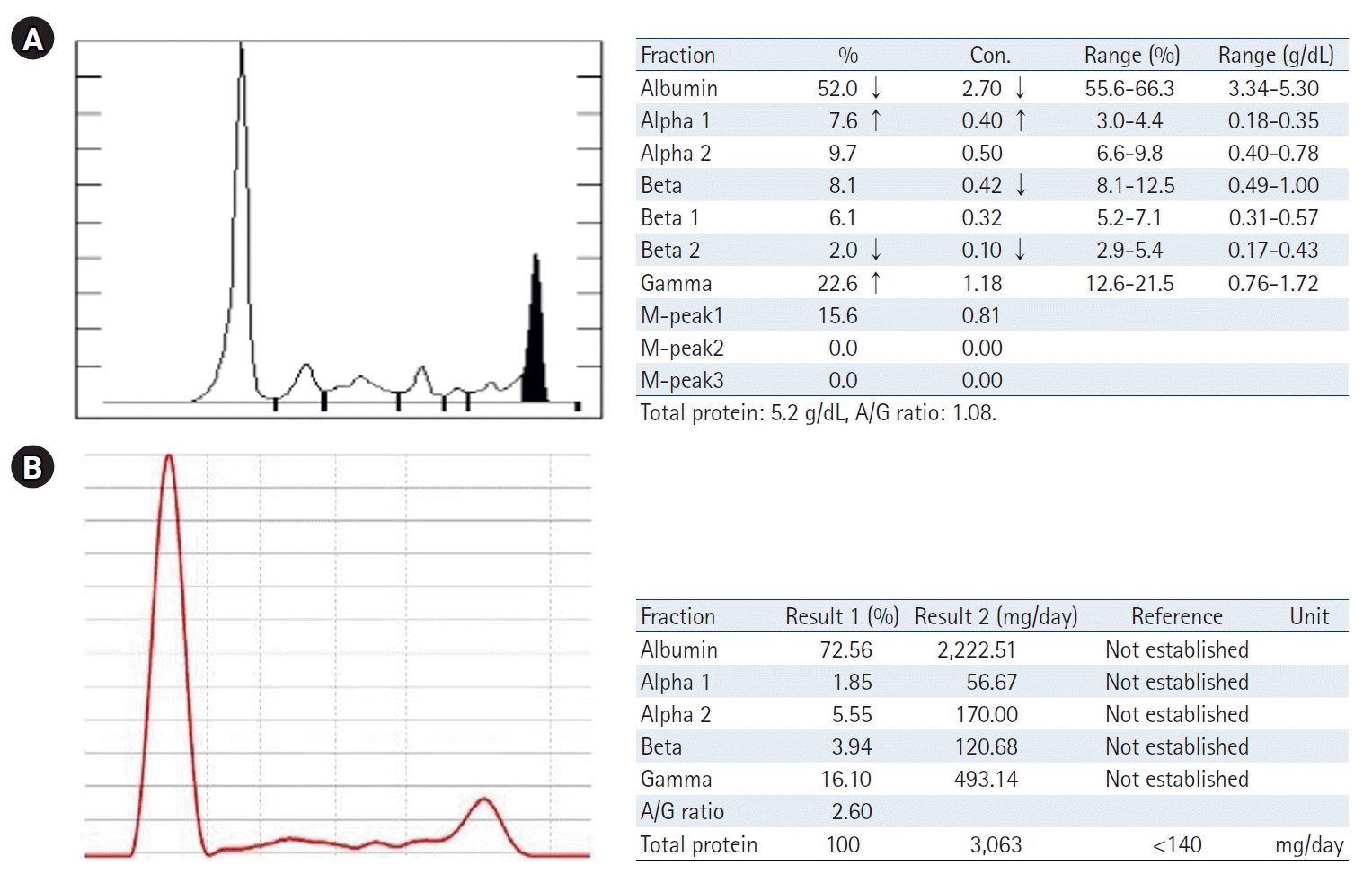
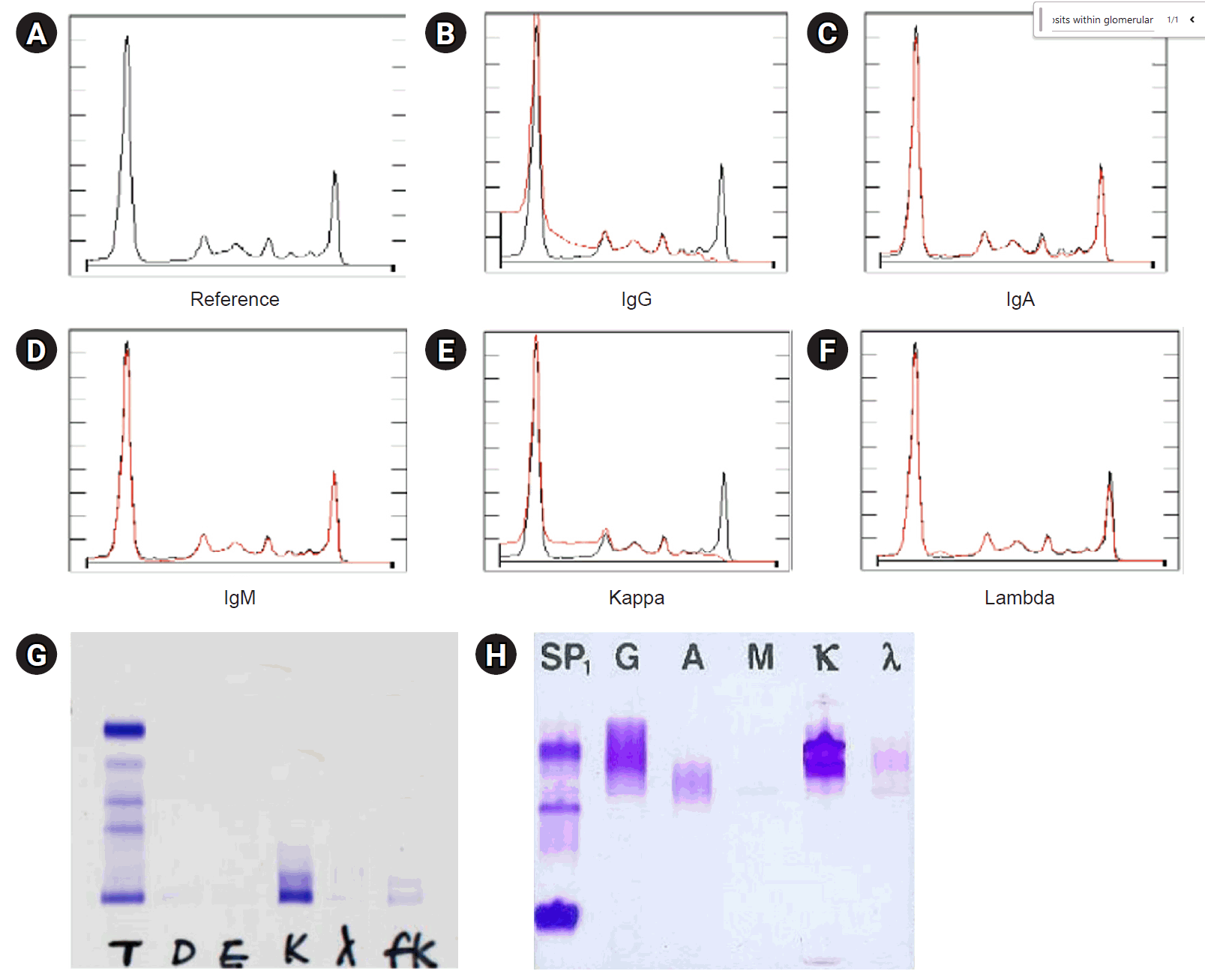
C3 and C4 levels were reduced, measuring 32.6 mg/dL and less than 2 mg/mL, respectively. The blood tests were positive for antinuclear antibodies (ANA), anti-myeloperoxidase antibodies (MPO Ab), and anti-glomerular basement membrane antibodies (GBM Ab). Serum protein electrophoresis indicated the presence of paraproteins in the gamma region (Fig. 1). Additionally, serum immunofixation identified an IgG kappa paraprotein (Fig. 2). The results of the Ig quantitation test were as follows: IgG at 1,317 mg/dL, IgA at 99 mg/dL, IgM at 48 mg/dL, free kappa light chain at 7,877 mg/L, free lambda light chain at 19.16 mg/L, and a free kappa to lambda ratio of 411.09.
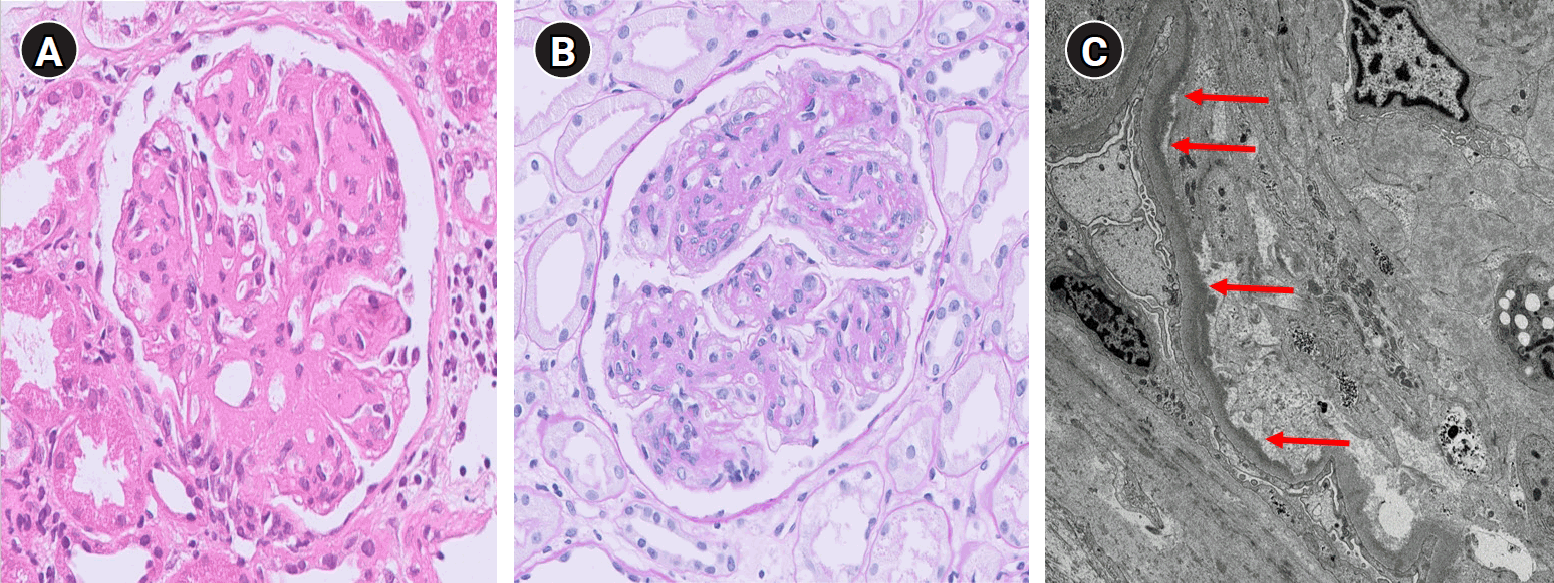
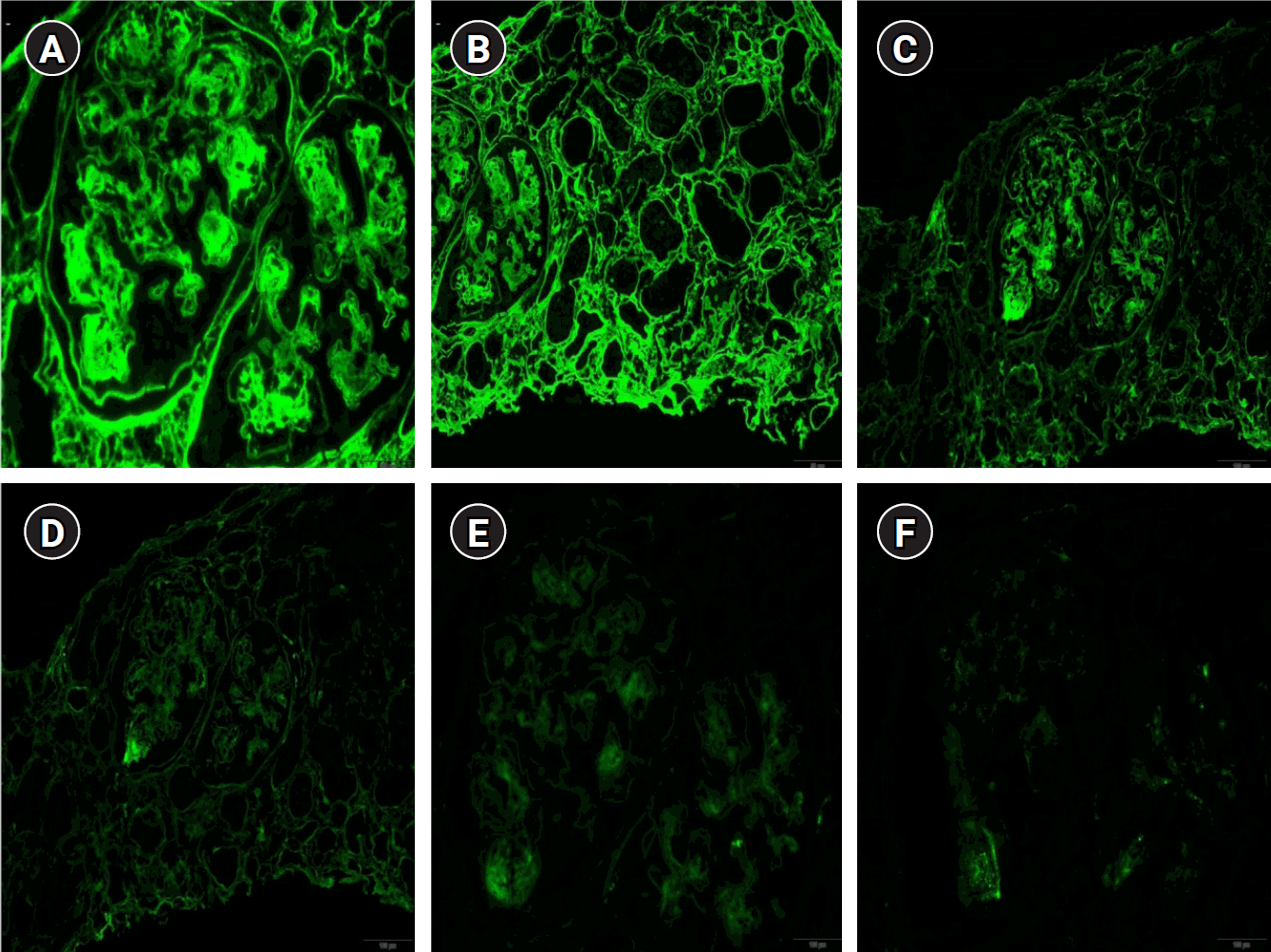
Considering the persistent proteinuria, decreased C3/C4 levels, and positivity for ANA, MPO, and GBM antibodies, it was necessary to exclude microscopic polyangiitis and anti-GBM disease. A renal biopsy revealed nodular glomerulosclerosis, severe mesangial expansion with marked hypercellularity, and fibrotic mesangial matrix expansion on light microscopy (LM) (Fig. 3). Immunofluorescence showed antibodies specific to the heavy chains of IgG and C3. IgG granular deposits (2+) were observed in the mesangium, and linear IgG staining (2+) was noted in the tubular basement membranes and along the peripheral capillary walls (Fig. 4). Electron microscopy (EM) revealed diffuse electron-dense “powdery” densities in the glomerular and tubular basement membranes (Fig. 3). Based on these findings, both microscopic polyangiitis and anti-GBM disease were excluded. Since amyloid was not detected under LM, amyloidosis was ruled out without the need for additional Congo red staining.
A bone marrow examination showed a marrow cellularity of 60% with widespread CD138-positive plasma cell proliferation, confirming the diagnosis of MM. The patient underwent a positron emission tomography-fluorodeoxyglucose (FDG) scan due to complaints of whole-body pain. This scan revealed diffuse heterogeneous FDG uptake in the bone marrow, indicative of myeloma involvement. Consequently, the patient was diagnosed with HCDD associated with MM (ISS stage III). Fluorescent in situ hybridization analysis did not detect del(17p) or t(14;16); however, trisomy 14 or amplification or translocation involving the Ig heavy chain (IgH) at 14q32 was suspected.
During the diagnostic process, dialysis was initiated due to persistent azotemia and pleural effusion, which did not respond to diuretics. After ruling out glomerulonephritis, the patient began treatment with the MM regimen, which includes bortezomib, lenalidomide, and dexamethasone. The treatment protocol involved subcutaneous bortezomib (2.21 mg on days 1, 4, 8, and 11), oral lenalidomide (5 mg daily from days 1 to 21), and intravenous dexamethasone (40 mg on days 1-4 and 8-11), administered over four cycles at monthly intervals. The only complication was a temporary discontinuation of lenalidomide for 8 days due to a skin rash during the first cycle. The patient reported no other significant issues in tolerating the medications.
Approximately 1 month after initiating anti-plasma cell therapy, the patient's kidney function improved, allowing the discontinuation of dialysis. Five months post-diagnosis, the patient underwent autologous peripheral blood stem cell transplantation and subsequently began lenalidomide therapy. As of September 2023, his laboratory results indicated a serum creatinine level of 0.64 mg/dL and a CKD-EPI eGFR of 109 mL/min/1.73 m2, which have remained stable.
Discussion
HCDD is a rare monoclonal Ig deposition disorder characterized by tissue deposits of truncated monoclonal IgH, typically associated with underlying plasma cell clones. Among the cases reported to date, γ-HCDD is the most frequently documented type, followed by α-HCDD and μ-HCDD [6]. α-HCDD is a form of extranodal marginal zone lymphoma of the mucosa-associated lymphoid tissue that affects the small intestine in young adults. It is associated with intestinal parasites and may respond to antibiotics in its early stages. However, if left untreated, it can progress to diffuse large B cell lymphoma. γ-HCDD is commonly associated with lymphoplasmacytic lymphoma. Its incidence is intermediate between those of α-HCDD and μ-HCDD, and it is characterized by extensive involvement of sites such as the bone marrow, lymph nodes, and spleen. μ-HCDD is a rare disorder with clinical characteristics resembling chronic lymphocytic leukemia. It is characterized by hepatosplenomegaly, vacuolated bone marrow plasma cells, free urinary light chains, and an abnormal serum mu chain [7,8].
Current evidence indicates that the loss of the first constant heavy chain domain (CH1 domain) results in the secretion of heavy chains from plasma cells prior to their association with light chains. Normally, heavy chains are retained in the endoplasmic reticulum due to the interaction between the CH1 domain and heavy-chain-binding proteins, and are only released following light chain binding [9,10]. Additionally, deletions in the variable regions of heavy chains modify the physicochemical properties of IgH, affecting their hydrophobicity and overall charge, which contributes to their deposition in tissues [11].
Common clinical manifestations include hypertension, progressive renal dysfunction, anemia, proteinuria, and hematuria. Laboratory results showed abnormalities in the serum-free kappa to lambda ratio and low complement levels, identified as distinctive features. During treatment, the patient’s complement levels normalized, suggesting a correlation between serum complement levels and disease activity in HCDD [12-14]. Regarding pathological findings, most cases exhibited a nodular glomerulosclerosis pattern on LM, and immnofluorescence (IF) revealed heavy chain deposits in the glomerular and tubular basement membranes. EM revealed fine, granular electron-dense deposits in the glomerular and tubular basement membranes.
HCDD can be diagnosed by combining histopathological findings with the analysis of serum or urine samples using electrophoresis and immunofixation. Histopathological findings, particularly immunofluorescence, are crucial for diagnosis. Oe et al. [6] suggested the following criteria: (1) monoclonal composition staining for a single class of Ig (γ, α, or μ) without corresponding light chains; (2) 2+ or greater intensity of staining; and (3) the presence of “powdery” electron-dense deposits within glomerular or tubular basement membranes on EM. In our case, pathological IF findings demonstrated strong positivity for antibodies against IgG heavy chains in the mesangium and tubular basement membranes, with no positivity for light chains. Additionally, a diffuse electron-dense “powdery” density was observed on EM, confirming the diagnosis of HCDD.
The treatment approach for HCDD varies depending on the subtype. For α-HCDD, which is often linked to infections, the initial treatment typically involves the use of antibiotics to address any concurrent infections, such as parasites. If symptomatic patients do not adequately respond to antibiotics, chemotherapy regimens used for non-Hodgkin lymphoma may be considered [7]. Based on limited empirical evidence, treatment of γ-HCDD is only indicated for symptomatic patients. In most reported cases, the patients received chemotherapy for MM treatment [6]. Major treatment regimens include bortezomib, lenalidomide, and dexamethasone [15]. The treatment for symptomatic patients with μ-HCDD closely resembles that for patients diagnosed with non-Hodgkin lymphoma [7].
The prognosis of γ-HCDD varies widely, from being an asymptomatic stable monoclonal heavy chain condition such as monoclonal gammopathy of undetermined significance to a rapidly worsening disease that may last only a few weeks, such as aggressive lymphoma. A review indicates that the median survival time is 7.4 years, with a range from 1 month to more than 21 years [16]. When MM is also present, patients can be assessed using MM risk stratification criteria. In our case, the patient is classified as standard risk due to the presence of trisomies and any one of the IgH translocations in cytogenetic abnormalities. It is recognized that in such cases, the negative prognosis associated with high-risk IgH translocations and del 17p may be mitigated [15].
In summary, HCDD is a rare disease with a generally poor prognosis. However, some cases have shown favorable outcomes when diagnosed and treated early. Therefore, improving the prognosis of HCDD requires recognizing its unique laboratory findings and pathological features and initiating treatment promptly.
 XML Download
XML Download