

 PDF
PDF Citation
Citation Print
Print



Introduction
Ectopic endometrium is a common chronic inflamma-tory disorder featured by the presence of growing glands and stroma from the endometrial tissue outside the uterine cavity. Although around 90% of females experience menstrual blood reflux, only a small proportion of this population develops endometriosis (1). A previous study suggested that ectopic lesions may originate from endometrial mesenchymal stem cells (MSCs) present in retrograde menstrual blood. However, ectopic MSCs (ect-MSCs) exhibited higher rates of proliferation, clone formation, invasion, and migration compared to eutopic MSCs (eut-MSCs). Furthermore, experiments conducted on mice demonstrated that ect-MSCs could invade surrounding tissues and promote neovascularization (2).
An increasing amount of evidence has demonstrated that the activation of the local inflammatory microenvironment contributes to the formation and progression of endometriosis (3). It is well-known that innate immunity serves as the first line of defense against pathogens in mammals and can rapidly recognize conservative pathogen-associated molecular patterns through pattern recognition receptors (PRRs). Among the PRR families, nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) are considered the primary intracellular PRRs. Notably, NOD1 and NOD2 represent crucial members of the NLR family (4). Abnormal expression of NOD1 has been impli-cated in various inflammatory diseases. In endometriosis peritoneal lavage fluid cells, increased mRNA expression of NOD1, NOD2, interleukin (IL)-1β, IL-6, IL-8, IL-10, interferon-γ, and tumor necrosis factor-α (TNF-α) was observed. Additionally, upregulated levels of NOD2 in endometrial stromal cells (ESCs) promoted the secretion of IL-6, IL-8, TNF-α, and monocyte chemoattractant protein-1 (5-7). Based on these findings, we hypothesized that NOD1 may play a role in the progression of endometriosis. Therefore, this study was aimed to investigate the differential functional expression of NOD1 between normal, eutopic, and ectopic endometrial MSCs.
Materials and Methods
Collection of tissues from normal, ectopic, and eutopic endometrium
Our study was approved by the Human Research Ethics Committee of Henan Provincial People’s Hospital (Zheng-zhou, China). Sixteen healthy females aged 32.46±5.96 years without prior medical interventions and 43 endometriosis patients aged 34.87±6.73 years were enrolled and the informed written consent was obtained. Exclusion criteria included females undergoing hormone treatment, those with abnormal uterine bleeding, submucous myoma, endometrial polyps, benign and malignant tumors, autoimmune diseases, diabetes, hypertension, and other serious systemic diseases. The ectopic and eutopic endometrium tissues were preserved in a mixture composed of 50% Dulbecco’s modified Eagle’s medium and 50% Ham’s F-12 medium (DMEM/F-12; HyClone) or fixed in 4% paraformaldehyde. A portion of the tissues was transported to the lab in DMEM/F-12, washed in phosphate buffer saline (PBS; HyClone), and used for primary cell isolation. The remaining tissues were fixed in paraformaldehyde for 24 hours and then embedded in paraffin for immunohistochemistry analysis.
Immunohistochemistry
The simplified immunohistochemistry protocol used is as follows: Paraffin sections (3 μm thick) of the ectopic and eutopic endometrium were dewaxed in xylene and ethanol, incubated in 3% hydrogen peroxide and goat serum albumin to remove endogenous peroxidase. The sections were incubated overnight with the following antibodies: 10 μg/ml rabbit anti-human vimentin, 10 μg/ml rabbit anti-human CK7, and 25 μg/ml mouse anti-human NOD1. PBS was used as a negative control. Subsequently, the sections were incubated within a moist chamber at the temperature of 4℃, followed by washing for 3 times. Next, the expression levels of proteins were determined utilizing AB complex (streptavidin/peroxidase) method (SP-9001; Zhongshanjinqiao). Color development was achieved using 3,3’-diaminobenzidine, followed by counterstaining with hematoxylin.
RNA extraction and real time quantitative pattern recognition receptor
RNA was extracted from both tissues and cells using TRIzol reagents (Invitrogen), followed by reverse transcription into cDNA using a PrimeScript RT reagent Kit (TaKaRa) in an Applied Biosystems pattern recognition receptor (PCR) machine (Applied Biosystems). The cDNA of each sample was diluted and amplified for real time quantitative PCR (RT-qPCR) in a final volume of 20 μl, containing 2 μl of the cDNA template, SYBR Premix Ex Taq II, and Rox reference dye (TaKaRa). The forward and reverse primers were purchased from Sangon Biotech. The primer pairs used for gene amplification of glyceral-dehyde-3phosphate dehydrogenase (GAPDH), NOD1, transforming growth factor-β1 (TGF-β1), vascular endothelial growth factor A (VEGFA), matrix metalloproteinase 2 (MMP2), MMP3, and IL-8 are as follow:
GAPDH: forward 5’-GCACCGTCAAGGCTGAGAAC-3’, reverse 5’-TGGTGAAGACGCCAGTGGA-3;
NOD1: forward 5’-TACTGAAAAGCAATCGGGAACT-3’, reverse 5’-GTAGAGGAAGAACTCGGACACC-3’;
TGF-β1: forward 5’-GGCCAGATCCTGTCCAAGC-3’,reverse 5’-GTGGGTTTCCACCATTAGCAC-3’;
VEGFA: forward 5’-CGGATCAAACCTCACCAAG-3’, reverse 5’-ACGCTCCAGGACTTATACC-3’,
MMP2: forward 5’-GCAATACCTGAACACCTTCTATG-3’, reverse 5’-ATTCTGGTCAAGATCACCTGTC-3’,
MMP3: forward 5’-GGTCTCTTTCACTCAGCCAACAC-3’, reverse 5’-CAGGCGGAACCGAGTCAGG-3’;
IL-8: forward 5’-ACTGAGAGTGATTGAGAGTGGAC-3’, reverse 5’-AACCCTCTGCACCCAGTTTTC-3’.
The reactions were performed at 95℃ for 30 seconds, followed by 40 cycles at 95℃ for 5 seconds and 60℃ for 30 seconds. Each sample was analyzed in triplicate using an ABI Prism 7500 Sequence Detector (Applied Biosystems). The amplified sequences were quantified against a standard curve. The expressions of the genes were normalized to that of GAPDH.
Primary cell isolation and culture
Stromal cells and MSCs were isolated from the eutopic endometrium and ectopic endometriosis tissues using collagenase II digestion (Sigma-Aldrich). The primary cells were seeded into DMEM/F-12 (Gibco) complete medium supplemented with 10% inactivated fetal bovine serum (FBS; Gibco), 100 U/ml penicillin, and 100 mg/ml streptomycin. The cells were incubated at 37℃ with 5% CO2. Non-adherent hematopoietic cells were removed by washing with PBS after 24 hours to isolate pure stromal cells. When the cells reached approximately 90% confluence, they were passaged using 0.25% Trypsin-ethylene diamine tetraacetic acid (EDTA; Gibco) at a 1:2 ratio. Differentiation studies were conducted on cells from passages 3 and 4.
Flow cytometry analysis and sorting
Stromal cells were seeded into 6-well plates until they reached 90% confluency. Subsequently, the cells were digested using 0.25% trypsin, centrifuged at 1,000 rpm for 5 minutes, and the supernatant was discarded. The cells were then washed twice with PBS and suspended in 1 ml of sheath solution. Afterwards, the cell concentration was adjusted to 1×106/ml. Next, 100 μl of the cell suspension was transferred to each tube and incubated with antibodies against CD31, CD44, CD73, CD140b, CD146, SUSD2, and CD271 for 30 minutes. Following the incubation, 500 μl of sheath solution was added. The antibody-labeled cells were subsequently analyzed and sorted using a FACSCalibur Flow Cytometer (BD Biosciences) within 1 hour. The obtained data was analyzed using FlowJo 10 software (FlowJo, LLC).
Identification of the differentiation characteristics of the MSCs
To evaluate the differentiation capacities of osteogenic, adipogenic, and chondrogenic phenotypes, three lineage differentiation detection kits (Cyagen) were utilized following the manufacturer’s instructions. The MSCs were seeded into a 24-well plate (Corning) with 500 µl of complete culture medium at a density of 5×104 cells per well. Once the cells reached 80% confluency, the complete medium was replaced with the differentiation medium. The medium was changed every 3 days over a period of 21 days. Staining with Oil Red O, Alizarin Red, and Alcian Blue dye solutions was performed to evaluate the adipocyte, osteoblast, and chondrocyte genetic capacity of the MSCs.
Clone formation assay
Cells were seeded into 6-well plates at a density of 1×103 cells per well. eut-MSCs and ect-MSCs were pretreated with various concentrations of NOD1 ligand γ-D-glutamyl-meso-diaminopimelic acid (Tri-DAP) (Invitrogen) or NOD1 inhibitor ML-130 for 10 days until obvious colony-forming unit (CFU) formation (more than fifty cells per CFU) was observed. Subsequently, the cells were fixed and stained using Giemsa’s stain (Solarbio).
Cells proliferation assay
To evaluate MSC proliferation, a cell counting assay kit (Cell Counting Kit-8, CCK-8; Tongren) and immunofluo-rescence for Ki67 were used. The CCK-8 kit utilizes a water-soluble tetrazolium salt that is reduced through cellular dehydrogenase activity to present a yellow formazan dye, in proportion to living cell number: the MSCs were seeded into 96-well plates at a density of 1×103 cells per well. eut-MSCs were pretreated with various concentrations of Tri-DAP for 72 hours, while ect-MSCs were pretreated with various concentrations of ML-130. After-ward, CCK-8 assay (10 μl) was supplemented to each well, followed by the incubation of the mixture for 2.5 hours. Subsequently, the optical density of each well was determined at 450 nm using an enzyme-linked immuno-sorbent assay (ELISA) Reader (Molecular Devices) and the number of cells were calculated. Ki67 immunofluo-rescence staining: The primary cells cultured on chamber slides were fixed using cold 4% paraformaldehyde and then permeabilized with 0.1% Triton X-100 for 15 minutes. After washing using PBS, the cells were incubated overnight with mouse anti-human Ki67 (diluted 1:200; ZSGB-BIO) as a primary antibodies, and then the slides were incubated with Alexa Fluorconjugated (green fluorescence) as secondary antibodies (Abcam) for another 1 hours at room temperature and then counter stained with 4’,6-dia-midino-2-phenylindole (blue color, P36966; Invitrogen). The stained cells were viewed using laser scanning confocal microscope (LSM780; Zeiss). The proliferative cell index was calculated as the percentage of green nucleus cells in each total cell number (green and blue nucleus staining cells).
Cell migration and invasion assays
MSCs were seeded into 12-well transwell plates (Corning) at a density of 5×104 cells in 200 μl per well and cultured with various concentrations of ML-130 (0.28 or 0.56 μM) for 24 hours. The cells on the lower membranes of the wells were then fixed and stained with Giemsa’s stain. The number of cells that had successfully migrated and invaded was calculated to analyze their migration and invasion capacities.
Cell apoptosis assay
Apoptosis Detection Kit I (BD Biosciences) was used to evaluate the ratio of apoptotic cells. The cells were seeded into 12-well plates (Corning) at a density of 1×106 cells per well and cultured with or without ML-130 for 24 and 48 hours. Cells were harvested using 0.25% trypsin without EDTA and collected with PBS into individual polystyrene round-bottom tubes 2045 (BD Falcon). Then, they were stained with Annexin-V and propidium iodide (PI) according to the manufacturer’s instructions simplified as follows: to each cell suspension, 10 μl Alexa Fluor 488-conjugated Annexin-V and 10 μl PI reagent were added. Cells were incubated in the dark for 15 minutes at room temperature. At the end of the incubation, the 400 μl binding buffer was added. Analysis was conducted using a fluorescence activated cell sorter (FACS) (FACSCalibur Flow Cytometer) as well as CellQuest software (BD Bioscie-nces). The percentage of early and late apoptotic cells was calculated using Annexin-V and PI double-positive cell staining, respectively.
Statistics
The statistical tests used in this study were identified unambiguously. The t-test with one or two tails, linear correlation in the parametric (Pearson) or non-parametric (Spearman) method were employed. Statistical analysis was carried out utilizing SPSS Statistics 19.0 software (IBM Corp.), while Prism 5.0 (GraphPad) was adopted for graph plotting. Quantitative variables between groups were compared using Student’s t-test (for normal distribution) or Mann–Whitney U-test (for non-normal distribution), and one-way or two-way ANOVA was used for multiple com-parisons. Pearson χ2 test or Fisher’s exact test was utilized to compare qualitative variables. Survival curves were generated using the Kaplan–Meier method and compared using the log-rank test. p<0.05 was considered statistically significant.
Results
Differentiation expression of NOD1 in ectopic and eutopic endometrium
To identify the ectopic and eutopic endometrium tissues, HE staining was performed (Supplementary Fig. S1A, S1E), followed by CK7 and vimentin staining to distinguish between endometrium and endometriosis. The endometrium exhibited diffuse glands in the stromal cells (Supplementary Fig. S1A-S1D), while the endometriosis primarily consisted of stromal cells (Supplementary Fig. S1E-S1H).
RT-qPCR and immunohistochemistry were employed to compare the mRNA and protein expression levels of NOD1 between ectopic and eutopic endometrium, respectively. The immunohistochemistry results demonstrated strong positive expression of NOD1 in the endometriosis (Fig. 1A-1D). Moreover, NOD1 mRNA expression was higher in the extopic endometrium (ect-EM) tissue/endometriosis with statistical significance compared to the eutopic endometrium (eut-EM) tissue/endometrium (ect-EM tissue and eut-EM tissue were paired samples and derived from the same patient) (Fig. 1E). Notably, for patients with endometriosis, NOD1 expression in their eut-EM tissue did not differ significantly from that observed in the endometrium (EN) from healthy females (Fig. 1E, EN vs. eut-EM, p=0.738).
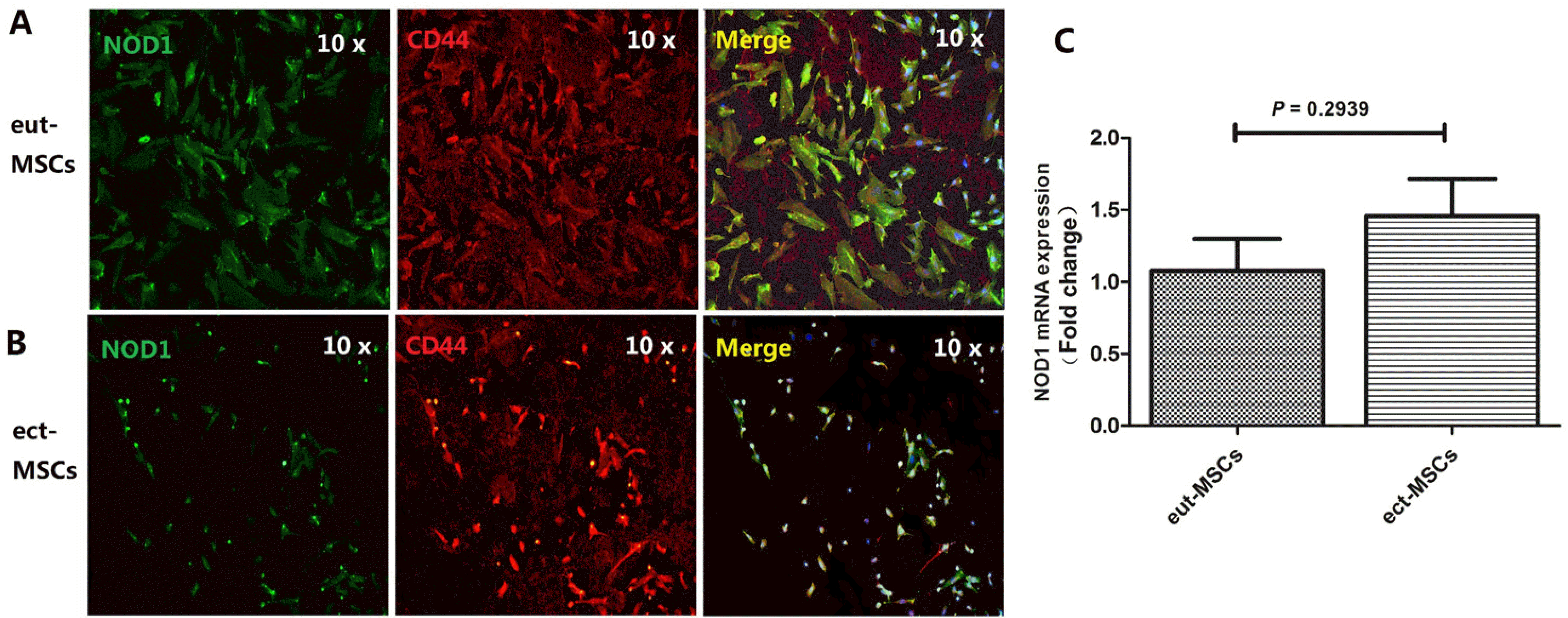
To explore whether and how NOD1 is functionally expressed in MSC-assisted endometrial ectopic implants, MSCs were isolated from the endometrial and endometriosis tissues. Adherent eutopic and ectopic ESCs were grown (Supplementary Fig. S2A, S2C). CD45(−) & CD31(−) & CD44(+) stromal cells were selected as MSCs, achieving a purity of approximately 98% after 72 hours of culture (Supplementary Fig. S2B, S2D). The eut-MSCs and ect-MSCs exhibited spindle-shaped or short and conical morphology. Adipogenic, osteogenic, and chondrogenic differentiation successfully induced multilineage differentiation potential of CD44(+) MSCs. Immunostaining revealed the presence of NOD1 protein in both CD44(+) eut-MSCs and ect-MSCs (Fig. 2A, 2B). Additionally, an increasing trend was observed in NOD1 mRNA expression among the ect-MSCs, although the trend was not statistically significant (Fig. 2C).
High NOD1 expression functionally affects the viability of eut-MSCs
Stimulation with the various concentration of NOD1 ligand, Tri-DAP, increased the number of CFUs and the proliferation of eut-MSCs (Fig. 3A, 3B), which was consistent with their increased viability and proliferation rates in Ki67 assays (Fig. 3C). The significantly higher proportion of proliferative cells were observed in the proliferation phase compared to the control cells (pretreated with PBS).
Since MSCs originating from endometrial tissue are implanted outside the uterine cavity, they need to tolerate the local microenvironment properly. To explore the differences in chemokine/cytokine profiles between eut-MSCs and ect-MSCs, we measured the transcription levels of IL-8 (Fig. 3D). The ect-MSC samples exhibited significantly higher IL-8 expression compared to the eut-MSC group (p<0.05). Moreover, the ect-MSC samples also performed significantly enhanced migratory capacity compared to the eut-MSC group (Supplementary Fig. S3) (p<0.0001).
Partial decrease in viability of ect-MSCs by ML-130
After pretreatment with ML-130, the inhibitory effect on ect-MSCs was evaluated at 24, 48, and 72 hours. Nota-bly, after 48 hours, the proliferation of ect-MSCs was significantly inhibited (Fig. 4A, right panel). In addition, clonogenesis, invasion, and migration abilities decreased following ML-130 pretreatment, as shown in Fig. 4A left panel and 4B. The transcription and secretion levels of IL-8 were higher in the ect-MSCs group with statistical significance compared to the eut-MSCs group (Fig. 4C, 4D). However, ML-130 pretreatment partially inhibited the upregulated expression of IL-8 (Fig. 4C, 4D).
Stimulation of primary ect-MSCs by ML-130 affects cell apoptosis
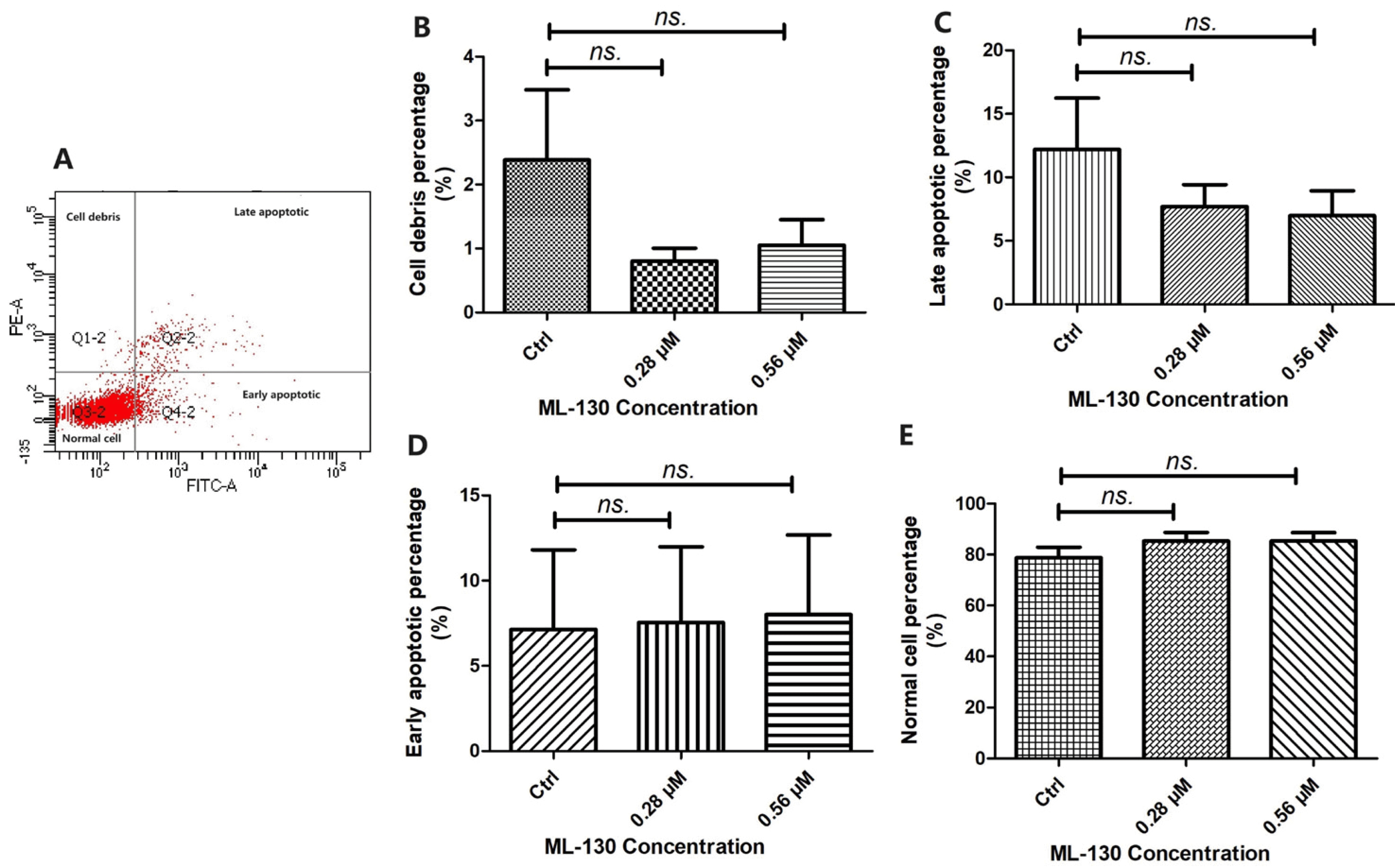
Primary ect-MSCs were stimulated with varying concentrations (0, 0.28, or 0.56 μM) of ML-130 for a duration of 48 hours (Fig. 5). Although there was a noticeable trend towards early apoptosis in the ect-MSCs, the observed difference between pretreatment cells and control cells were not statistically significant (Fig. 5B-5E) (p>0.05).
Discussion
NOD1, the first intracellular PRR identified in the NOD-like receptor family, is capable of recognizing degradation products of bacterial cell wall peptide glycans. It plays a crucial role in promoting the release of proinflammatory factors and participating in the innate immune response. Our study discovered higher mRNA expression levels of NOD1 in the ect-EM group compared to the eut-EM group. The components of the ect-EM need to tolerate their microenvironment. Previous research has demonstrated that the proliferation and invasion of ectopic ESCs (ect-ESCs) are promoted by MMP2 and MMP9, leading to the formation of ectopic lesions (8). Overexpre-ssion of VEGFA in ect-ESCs enhances their invasive and migratory capabilities. However, inhibiting VEGFA expression can counteract these effects. Placental and fetal membrane expression of NOD1 stimulates the secretion of IL-1 and IL-6 through activation of the RIP2-NF-κB/MAPK signaling pathway (9). Our study revealed that NOD1 could upregulate the IL-8 secretion level in cervical cancer cells (10). Other studies have reported higher levels of IL-8 secretion by peritoneal macrophages from patients with endometriosis (11). Elevated IL-8 expression is implicated in the adhesion and implantation of ectopic lesions (12, 13). Moreover, our findings demonstrated signi-ficantly higher IL-8 mRNA expression in ect-MSCs compared to eut-MSCs, suggesting that abnormal IL-8 expre-ssion in ect-MSCs may contribute to the development of ectopic endometriosis. Notably, ML-130, an inhibitor of NOD1, can partial decrease IL-8 upregulation and may functionally inhibit the formation of endometriosis in vitro.
Studies have identified elevated NOD1 expression at the mRNA level in endometriosis peritoneal lavage fluid cells (14). Our study focused on NOD1 expression in ect-MSCs obtained from endometriosis patients. The high expression of NOD1 in these ect-MSCs suggests their involvement in the development of ectopic endometriosis. Chlamydia trachomatis infection in vivo relies on NOD1-RIP2 to induce IL-8 expression, which contributes to the inflammatory reaction (15). Navarro et al. (16) demonstrated that NOD1 agonist induced the expressions of IL-6 and IL-8 in cerebral pericytes, thus promoting a perivascular inflammatory reaction through the NOD1-p38MAPK/NF-κB-IL-8 signaling pathway.
MMP2 and MMP3 can hydrolyze various components of the extracellular matrix and basement membranes such as fibronectin, laminin, collagen IV, and gelatin. This hydrolysis can result in the disruption of the basement membrane and extracellular matrix, allowing the eutopic endometrium to breach the local barrier and promote the ectopic growth of adjacent tissues (17, 18). Multiple studies have reported higher expression of MMP3 in eutopic endometrium compared to normal endometrium. Muharam et al. (19) detected down-regulated levels of MMP3 expression in normal endometrium tissues than in eutopic endometrium, while Luddi et al. (17) found lower expression of MMP3 in endometrium. Our study was not consistent with above findings, revealing significantly higher transcription levels of MMP2 and MMP3 in eut-MSCs compared to ect-MSCs (data was not shown). This difference may be attributed to the fact that most participants in our study were in the proliferative phase with low progesterone levels, which can inhibit the expression of MMPs in the endometrium (20).
Furthermore, we investigated the impact of NOD1 expression on the invasion and migration abilities of ect-MSCs. Our findings indicated that ML-130 partially hindered the invasion and migration abilities of ect-MSCs, suggesting the involvement of NOD1 in their invasion and migration. The development of ectopic lesions relies on an imbalance between the expression levels of pro-apoptotic and anti-apoptotic genes in ESCs (21). Delbandi et al. (22) discovered that the apoptotic ability of ectopic ESCs was lower compared to eutopic ESCs. Previous research has indicated that NOD1, a pro-apoptotic protein, can induce apoptosis by interacting with caspase or apaf1, while NF-κB inhibits apoptosis. NOD proteins can activate both NF-κB and caspase signaling pathways (23). Nevertheless, in our study, the apoptosis rate of the ect-MSCs was not determined along with the inhibition of NOD1 expression. Liu et al. (24) found that ect-MSCs exhibited a higher level of proliferation, while Burlev et al. (25) observed no difference in the proliferation abilities between ect-MSCs and eut-MSCs. Overall, our study uncovered that the NOD1 ligand Tri-DAP can promote the proliferation of eut-MSCs. Conversely, ML-130 inhibits the proliferation and migration/invasion of ect-MSCs. However, further exploration is required to elucidate the underlying signaling pathway.
Supplementary Materials
Supplementary data including three figures can be found with this article online at https://doi.org/10.15283/ijsc22200

 XML Download
XML Download