

 PDF
PDF Citation
Citation Print
Print



Introduction
The tumor microenvironment (TME) comprises various cell types and is critical for tumor progression, relapse, metastasis, and therapy resistance (1). Immune cells, fibroblasts, endothelial cells, and nerve cells act as pro-tumor and anti-tumor stromal cells. Furthermore, non-cellular components, including the extracellular matrix (ECM), metabolites, hypoxia, and acidity, contribute to tumor development (1, 2).
Cancer stem cells (CSCs) are a minor subpopulation of cells that are considered the most crucial components of the TME for tumor development. CSCs were initially discovered in acute myeloid leukemia in 1994 (3). In solid tumors, breast cancer stem cells (BCSCs) were the first characterized CSCs as CD44+CD24−/low population (4). CSCs exhibit self-renewal, unlimited proliferation, and dormant-state maintenance that are affected by the TME. Because CSCs may contribute to cancer recurrence or metastasis, understanding the mechanisms that regulate the activity and life cycle of CSCs in the TME is important for developing effective tumor treatment strategies.
T cells drive immune responses by counteracting specific pathogen-derived antigens. During antigen recognition, T cells differentiate into multiple types of effector cells that confer immunological protection to the host (5). However, in the TME, tumor-specific T cells tend to be dysfunctional because most tumor antigens are immuno-ignorant self-molecules (6). Regardless of the ability of T cells to kill tumors, the TME accumulates T cell subsets that encourage tumor growth and inhibit effector T cell differentiation through the action of diverse components of the tumor bed (7). Regulatory T cells (Tregs) are among the most potent T cells that dampen effector cell function. Tregs maintain immune homeostasis in a normal state, but they are excessively enriched in the TME and are associated with defective effector cell responses and, in turn, poor prognosis in patients with malignancy (8). Interle-ukin (IL)-17-secreting CD4+ T (Th17) cells also promote tumor progression by enhancing the production of vascular endothelial growth factor (VEGF), prostaglandin E2 (PGE2), and nitric oxide from tumor and stromal cells, which leads to angiogenesis, although the function of Th17 in the TME remains controversial (9, 10).
Glutathione (GSH) is a tripeptide made up of glutamate, cysteine, and glycine and is a major intracellular antioxidant that exists at a concentration of mM (11). GSH reacts with most reactive oxygen species (ROS) such as H2O2, superoxide, and hydroxyl radicals, thereby oxidizing itself. GSH is then reduced by the electrons derived from reduced nicotinamide adenine dinucleotide phosphate (NADPH), generating a redox buffering cycle. This cycle not only removes ROS from cells but also regulates various redox signaling pathways by modulating the oxidation and reduction of cysteine residues of proteins, including tumor growth and therapy resistance. Research on developing real-time GSH monitoring tools within live cells has revealed that GSH is dynamically regulated within cells and is a critical factor in controlling the stemness and the-rapeutic potency of stem cells (12-15).
In this review, we comprehensively discuss how the GSH dynamics in CSCs and T cells within the TME induce and regulate tumor development. Furthermore, we highlight the recently discovered therapeutic strategies that target GSH dynamics.
CSCs and GSH Dynamics in TME
CSCs and GSH in tumor heterogeneity
Technical advances in nucleotide sequencing and molecular analyses have made tumor analysis more precise, revealing that tumors occur and evolve with intertumoral and intratumoral heterogeneity (1). Intertumoral heterogeneity is caused by various cancers, resulting in different tumor characteristics for each patient. In contrast, intratumoral heterogeneity arises from the dynamic development of cancer because of genomic instability and clonal evolution/selection and is a major factor that complicates cancer treatment. CSCs are considered the primary cells contributing to intratumoral heterogeneity owing to their characteristics of dissemination and resistance to therapy. CSCs are heterogeneous, with different markers depending on the organ in which they arise (Table 1).
Metabolic reprogramming, which depends on the TME, is crucial for tumor survival (2). Cancer cells tend to localize mostly under oxidative stress owing to the high levels of ROS generated by oncogenic signals, hypoxia, and activated immune cells. ROS are required for tumor development, including activation of proliferation signal transduction and gaining additional genetic mutation hits; however, excess ROS are detrimental because they damage macromolecules, including DNA, proteins, and lipids. Antioxidant systems are required to adjust the redox balance in tumors for survival and propagation. GSH is a major tumor antioxidant that removes excessive ROS and regulates tumor redox signals. In addition, aging is a crucial cause of tumor development that decreases cellular GSH levels (16). The levels of GSH vary and dynamically change among individual cancer cell lines (17) and CSCs (Table 1) (18-24) in response to the TME, favoring tumor development. Therefore, tumor GSH heterogeneity should be considered an important factor for a complete understanding of tumor development.
GSH dynamics in CSCs
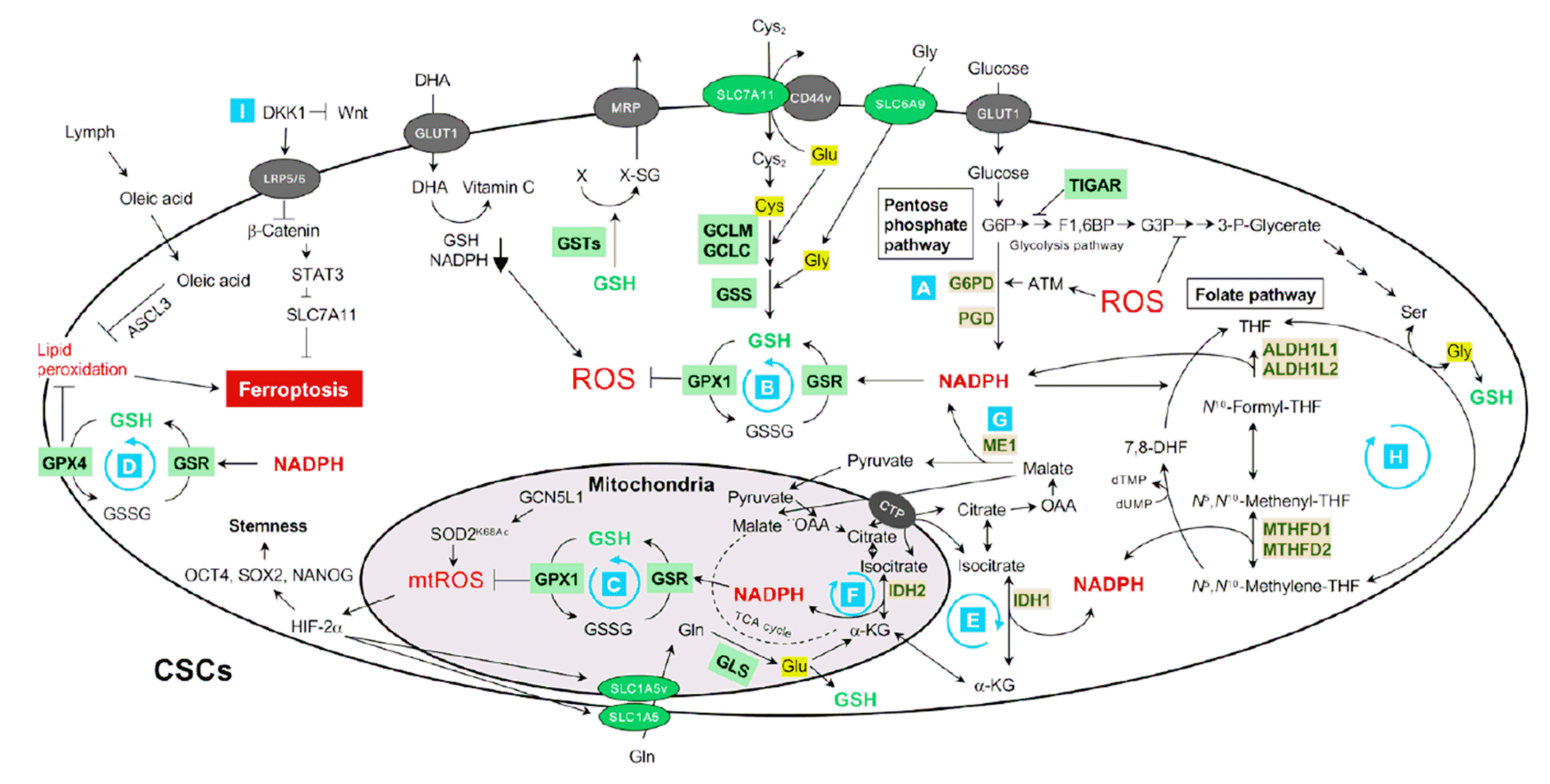
In CSCs, GSH is dynamically regulated by synthesis and regeneration cycles (Fig. 1). GSH is L-γ-glutamyl-L-cysteinyl-glycine synthesized by glutamate-cysteine ligase (GCL) and glutathione synthetase (GSS) in the cytosol. GCL is a heterodimeric enzyme composed of two subunits: the glutamate-cysteine ligase catalytic subunit (GCLC) and the glutamate-cysteine ligase modifier subunit (GCLM), which catalyze a rate-limiting reaction in GSH synthesis. As cytoplasmic cysteine levels are tightly controlled owing to cysteine toxicity (25), cysteine must be maintained at low concentrations within cells and transported from outside the cells through SLC7A11 (also referred to as xCT), a cystine-glutamate antiporter, for GSH synthesis. SLC7A11 is stabilized by its interaction with CD44 variants (CD44v), which are expressed in CSCs derived from the colorectum (26), stomach (27), and bladder (28), leading to increased intracellular GSH levels (Table 1). Glutaminase (GLS) produces glutamate from glutamine, which is then transported into the cells through SLC1A5, a sodium-dependent neutral amino acid transporter. Glycine can be supplied by SLC6A9 (also referred to as sodium- and chloride-dependent glycine transporter 1) and de novo synthesized from 3-phosphoglycerate, an intermediate of glyco-lysis. The glycine synthesis can be regulated by p53. TP53-inducible glycolysis and apoptosis regulator (TIGAR) is p53-dependently expressed, inhibits glycolysis through fructose-2,6-bisphosphatase activity, and decreases glycine synthesis. However, TIGAR activates the pentose phosphate pathway (PPP), which is the main route of NADPH production, and decreases ROS production and cell death (Fig. 1A) (29). Moreover, under hypoxic conditions, TIGAR is localized to the mitochondria and activates hexokinase 2 by directly binding to decrease mitochondrial ROS (mtROS) and protect cells from apoptosis (30). Furthermore, p53 indirectly increases GSH synthesis under serine and glycine starvation conditions, as p53 arrests the cell cycle by inducing p21, and de novo-synthesized glycine is utilized for GSH, but not purines (31).
GSH removes peroxides by reacting with glutathione peroxidase (GPX), which is encoded by eight different isozymes (GPX1-8) on human chromosomes. Human GPX1, GPX2, GPX3, GPX4, and GPX6 utilize selenocysteine as their active site. GPX1 inactivates cytoplasmic and mitochondrial peroxides (Fig. 1B, 1C) and GPX4 removes membrane lipid peroxides (Fig. 1D), oxidizing GSH into glutathione disulfide (GSSG, an oxidized GSH). GSH can be regenerated by glutathione reductase (GSR) using NADPH as an electron donor to generate a redox-buffering cycle (Fig. 1B-1D). NADPH can be supplied by the enzymatic reactions of: (1) glucose-6-phosphate dehydrogenase (G6PD) and 6-phosphogluconate dehydrogenase (PGD) in PPP (Fig. 1A); (2) isocitrate dehydrogenase 1 and 2 (IDH1/2) (Fig. 1E, 1F); (3) malic enzyme 1 (ME1) (Fig. 1G); and (4) 10-formyltetrahydrofolate dehydrogenase 1 and 2 (ALDH1L1/2) and methylenetetrahydrofolate dehydrogenase 1 and 2 (MTHFD1/2) in the folate pathway (Fig. 1H) (32, 33).
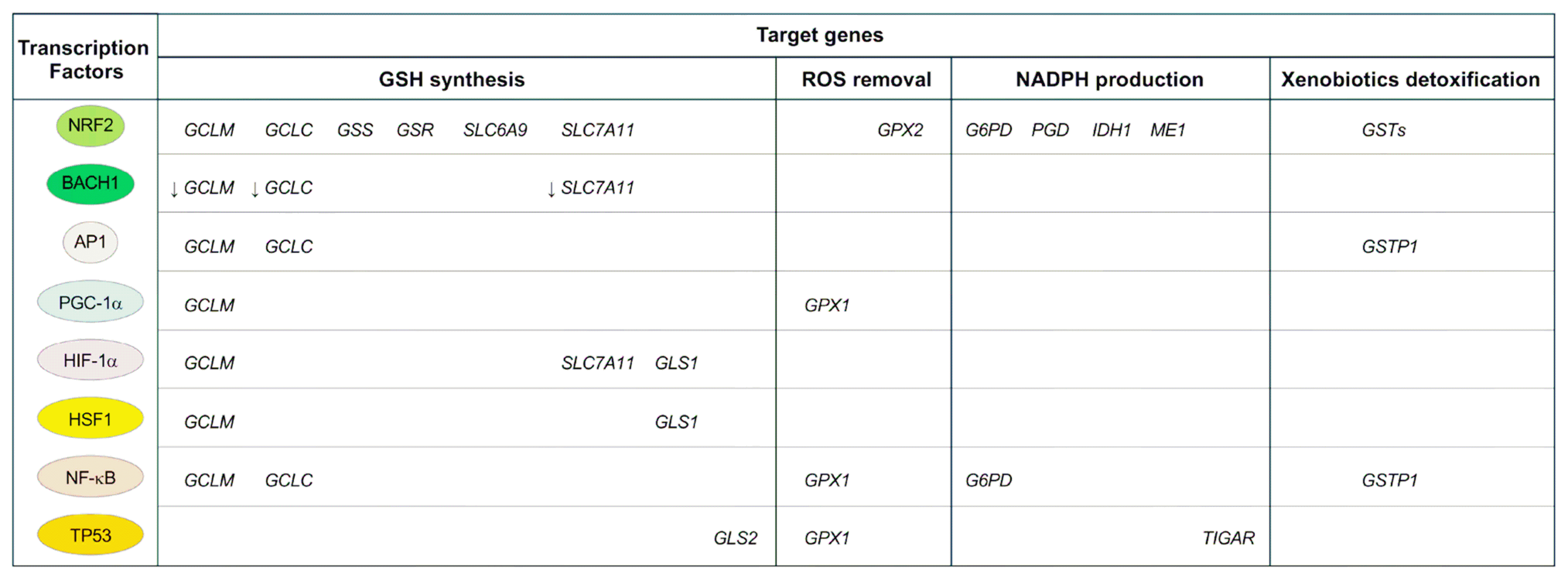
The GSH redox buffering system is regulated by various transcription factors (TFs) (Fig. 2). Nuclear factor erythroid 2-related factor 2 (NRF2) is the master TF for genes that synthesize GSH and generate NADPH. Under normal conditions, NRF2 is blocked by binding to Kelch-like ECH-associated protein 1 (KEAP1) and subsequent degradation in the cytosol via the ubiquitin-proteasomal system. Oxidative stress oxidizes the redox-sensitive cysteine residues of KEAP1, releasing NRF2. Free NRF2 moves into the nucleus and binds to antioxidant response elements located in the promoters of genes encoding the GSH redox buffering system.
Regulation of CSC stemness by GSH dynamics
The capacity of CSCs to self-renew, metastasize, and tolerate cancer therapy is regulated by cellular redox systems including GSH. BCSCs are reprogrammed by mitochondrial superoxide dismutase acetylation of lysine 68 (SOD2K68Ac)-promoted mtROS, which induces the expression of OCT4, SOX2, and NANOG through the stabilization of HIF-2α, favoring the upregulation of their stemness (Fig. 1) (34). HIF-2α also induces the expression of SLC1A5, a mitochondrial glutamine transporter, leading to increased GSH synthesis and drug resistance (Fig. 1) (35). In contrast, Lu et al. (36) demonstrated that chemotherapy increased the expression levels of SLC7A11 and GCLM in an HIF-1α-dependent manner, leading to the upregulation of GSH synthesis and NANOG expression in BCSCs.
The GSH redox buffering system is important for cancer cell metastasis. Disseminated cells exhibit reduced glucose uptake, leading to increased oxidative stress-induced cell death (37, 38). Oncogenes increase glucose uptake and NADPH generation through the PPP, leading to decreased ROS levels and increased cell survival (39). In KRAS-driven lung cancer, N-acetyl cysteine (NAC), a known precursor or mimetic of GSH, induces the transcriptional activation of BACH1-dependent hexokinase 2 and glyceraldehyde 3-phosphate dehydrogenase, promoting glycolysis-induced metastasis (40). NAC treatment also promotes melanoma metastasis, and metastasizing melanoma cells require metabolic changes with the induction of folate pathway enzymes, including ALDH1L1/2 and MTHFD1/2, for NADPH generation (Fig. 1H) (33). As ECM detachment, which is the initial process of cancer metastasis, increases intracellular ROS levels, cancer cells rewire their redox metabolism for metastasis (41). During redox reprogramming, the generation of mitochondrial NADPH by IDH1 and IDH2 is critical for cell growth without anchorage, suggesting that regeneration of mitochondrial GSH is vital for CSC proliferation during metastasis (Fig. 1C, 1F) (42). Melanoma cells metastasized through blood are susceptible to GPX4 inhibition-mediated ferroptosis, but melanoma cells through lymphatic vessels do not result from high levels of GSH and oleic acid in lymph fluid that inhibit lipid peroxidation (43). Furthermore, DKK1 (dickkopf-related protein 1) secreted by BCSCs inhibits the WNT signaling pathway and induces SLC7A11 expression, protecting BCSCs from ferroptosis (Fig. 1I) (44).
Targeting CSCs by controlling GSH dynamics
The GSH redox buffering system appears to have different and complicated effects on tumorigenesis depending on the tumor stage. In the initial stage of tumorigenesis, the GSH system is protective because the genetic mutation rate is increased, and ROS can potentiate cellular growth signaling pathways by inhibiting phosphatases such as PTEN (Fig. 3A) (45). However, in the later stages of tumorigenesis, treatment with antioxidants that compensate for GSH promotes metastasis of breast cancer, lung cancer, and melanoma by increasing the expression of metastatic genes and the survival rate of CSCs (33, 40, 43, 46). In clinical trials, dietary supplementation with antioxidants does not typically decrease cancer occurrence (47), but is associated with higher rates of lung and prostate cancer occurrence and mortality (48-50). These results suggest that GSH depletion is beneficial for tumor treatment.
GSH can be depleted by treatment with buthionine sulfoximine (BSO), an inhibitor of GCL, which is the rate-limiting step in GSH synthesis. However, BSO showed not only liver toxicity (51) but also resistance in most cancer cell lines (52), implying that GSH is dispensable owing to compensatory pathways. The thioredoxin (TRX) system has been proposed as an alternative to antioxidant pathways. Inhibition of both the GSH and TRX systems results in the synthetic lethality of cancer cells (52). BCSCs are also sensitive to combined targeting of the GSH and TRX pathways (Fig. 3B) (46). Deubiquitinase (DUB) pathway is another compensating pathway that protects cancer cells from ER and proteotoxic stress upon inhibition of GSH synthesis (53). Thus, blocking both the GSH and DUB pathways synergistically inhibits tumor growth.
NADPH is essential for GSH regeneration during redox buffering NADPH can be generated by G6PD, PGD, ME1, IDH1/2, ALDH1L1/2, and MTHFD1/2 (Fig. 1A, 1E, 1E-1H). Methotrexate, a dihydrofolate dehydrogenase inhibitor, ameliorates melanoma metastasis by inhibiting NADPH generation via the folate pathway (Fig. 1H) (33); however, it also blocks the synthesis of dTMP and glycine, a precursor of GSH. In contrast, dietary folate supplementation facilitates the advancement and worsening of breast cancer (54, 55), possibly by contributing to NADPH production. Ascorbate exerts anti-cancer effects by depleting GSH and NADPH in cancer cells (Fig. 1) (56). Dysregulation of GSH systems is also associated with iron-dependent lipid peroxidation, leading to the ferroptotic cell death of CSCs during tumor development (Fig. 1D) (43, 44). Indeed, SLC7A11 inhibitors such as erastin and sulfasalazine promote ferroptosis in CSCs by downregulating GSH synthesis (57). GPX4 and NADPH-generating enzymes are promising targets for ferroptosis in CSCs (Fig. 1) (58). GLS is another possible target for controlling GSH dynamics because GLS inhibition reduces the glutamate and cysteine levels required for GSH synthesis (Fig. 1) (59). CB-893, a GLS1 inhibitor, was developed to KEAP1 mutant lung cancer (60).
Potential Role of GSH Dynamics in Modulating T Cells in the TME
Basic understanding of T cell activation
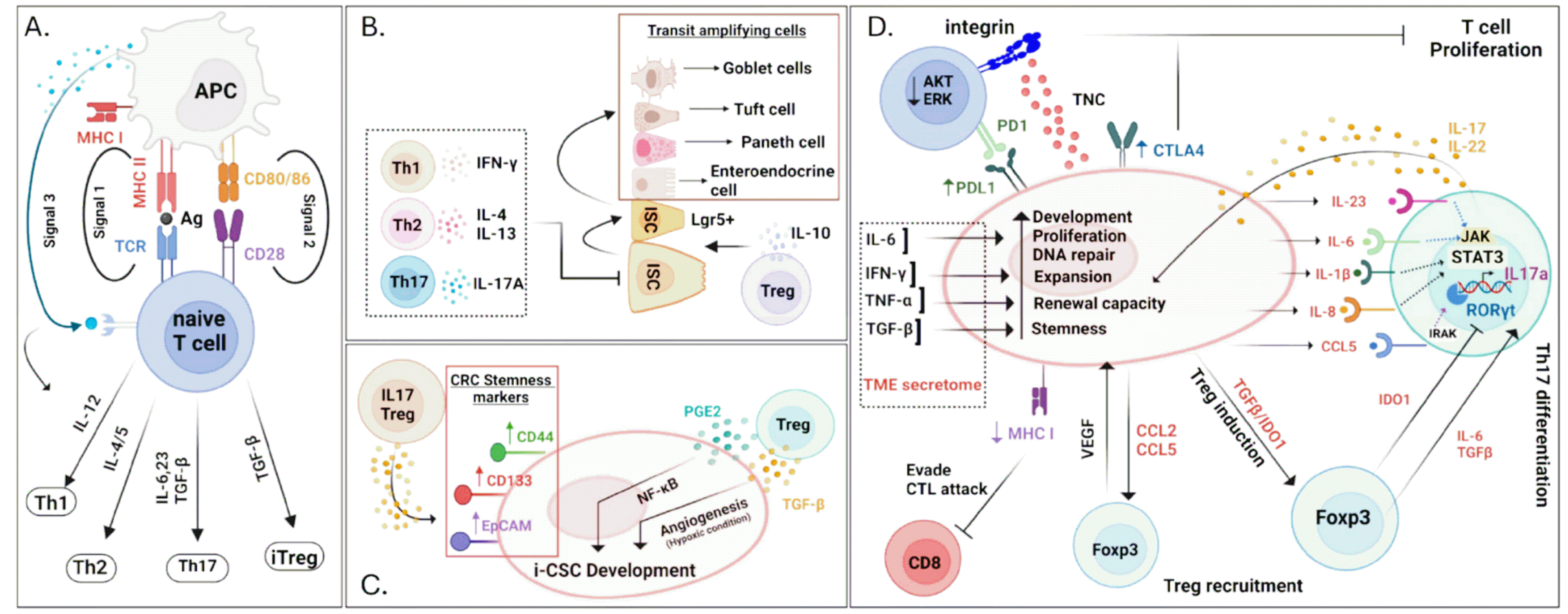
T cells are integral to adaptive immunity and crucial in protecting the host against pathogens and in exacerbating immune-related diseases (Fig. 4A). For full activation, T cells receive signals 1, 2, and 3 via the cell surface receptors. Signal 1 is initiated by the T cell receptor (TCR), which recognizes the complex of antigenic peptides and major histocompatibility complex classes I and II (MHC I and MHC II) of antigen-presenting cells (APCs). Signal 2, often referred to as co-stimulation, is essential for the complete activation of T cells by fortifying signal 1. This signal is typically generated by the ligation of co-stimulatory ligands on APCs with their receptors on T cells. One of the most well-studied pairs of molecules involved in this process is CD28 on T cells, and CD80 (B7-1) or CD86 (B7-2) on APCs. The binding of CD28 to CD80 or CD86 provides critical survival signals to T cells, leading to an increased expression of IL-2 and its receptor, IL-2R, to drive T cell proliferation. Signal 3 in T cell primarily involves the action of cytokines that direct the differentiation and functional maturation of T cells. After a T cell receives signal 1 through TCR engagement with the peptide-MHC complex and signal 2 through co-stimulatory interactions (such as CD28 binding to CD80/CD86), signal 3 determines the type of immune response that develops by influencing the T cell differentiation pathway. Signal 3 is defined by the cytokine environment surrounding the T cells during initial activation. Different cytokines promote the differentiation of T cells into various subsets of effector cells, each tailored to combat specific pathogens or regulate immune reactions. For instance, IL-12, secreted by APCs and other immune cells in response to certain bacterial or viral infections, promotes T helper 1 (Th1) differentiation. Th1 cells are particularly effective against intracellular pathogens and are involved in activating macrophages and inducing interferon (IFN)-γ production. IL-4, produced by APCs and other T cells, drives the differentiation of T cells into Th2 cells. Th2 cells are essential for combating extracellular parasites and play a critical role in allergic responses, primarily through the production of cytokines, such as IL-4, IL-5, and IL-13. Transforming growth factor-β (TGF-β) and IL-6 are important for the differentiation of T cells into Th17 cells. Th17 cells are important for the defense against fungal and bacterial infections and are involved in inflammation and autoimmunity. Tregs are induced by TGF-β, which maintains immune tolerance. Collectively, the regulation of these signals is vital for the functioning and balance of the immune system, which restricts host-damage threats.
T cell-stem cell axis in the TME
Crosstalk between T cells and intestinal stem cells: Unlike the universal expression of MHC I, MHC II is expressed only on APCs such as dendritic cells (DCs). Given that the TCRs of CD4+ T cells sense antigenic peptides loaded onto MHC II molecules, the biology of CD4+ T cells have been closely linked to APCs. Recently, however, using single-cell RNA sequencing, Biton et al. (61) found that a subset of Lgr5+ intestinal stem cells (ISCs) expresses MHC II molecules and that CD4+ T cells recognize and respond to antigens presented by ISCs (Fig. 4B). This observation indicates that MHC II on ISCs is functionally intact and that T cells and their cytokines can influence ISCs. Specifically, they demonstrated inflammatory cytokines like IFN-γ and IL-17 are linked to promoting differentiation processes, whereas IL-10 from Tregs tends to encourage stem cell maintenance. These interactions significantly affect the cellular composition of the intestinal lining under both normal and inflammatory conditions. Moreover, experimental manipulations that alter T cell dynamics or MHC II expression in ISCs have demonstrated shifts in epithelial dynamics and cell fate during immune responses, highlighting the dual role of ISCs in tissue regeneration and immune function.
Crosstalk between T cell and CSCs: Compelling research has thoroughly demonstrated how tumor masses interact with T cells; however, studies have only recently begun to clarify the interactions between T cells and CSCs in the TME. Notably, the network between Tregs and CSCs has been extensively investigated because Tregs play an integral role in the TME, as they have multiple arms not only for suppressing effector T cells but also for accelerating tumor growth/metastasis (62). In general, CSCs promote the accumulation of Tregs both directly and indirectly. CSCs actively attract Tregs by secreting chemokines CCL2 and CCL5 in a preclinical glioblastoma model (Fig. 4D) (63). Tregs migration by CCL2 was reliant on the presence of CCR4, as tumor infiltration is reduced in mice lacking CCR4. In a human ovarian cancer cell line, elevated levels of CCL5 facilitated Treg migration through a CCR5-dependent mechanism (64). CCL2 and CCL5 are also essential for myeloid cell migration, which indirectly facilitates Treg infiltration into the TME (63, 65). Furthermore, CSCs derived from multiple cell lines of cancer show increased levels of indoleamine 2,3-dioxygenase 1 and TGF-β, which enhances the recruitment and development of Tregs (66, 67). On the other hand, Tregs also benefit CSCs development. Tregs secrete PGE2, which accelerates the growth and migration of CSCs in a colorectal mouse model via the activation of NF-κB (Fig. 4C) (68). In the hypoxic TME, Tregs produce a significant amount of VEGF, which mediates angiogenesis and enhances the expansion of CSCs (69).
IL-17 released by Th17 cells increases the self-renewal capabilities of CSCs in various tumor models (70). Other Th17 cytokine, IL-22, also triggers STAT3 phosphorylation in tumor cells (71). STAT3 may serve as a crucial element in regulating the stemness and expansion of CSCs by maintaining their stem-like characteristics and the expression of stemness-associated genes, including Sox2, Nanog, and Oct4 (72). A recent study on the role of Tregs in colorectal cancer showed that Tregs express IL-17 under hypoxic conditions, and these IL-17+ Tregs contribute to the expansion of CSCs by upregulating stemness markers, such as CD44, EpCAM, and ALDH (Fig. 4C) (73).
CSCs evade the immune system by increasing the expression of immune checkpoint ligands. In multiple cancers such as gastric, breast, malignant mesothelioma, and bladder, there is a correlation between the expression of PD-L1 and CSC markers in aggressive tumors (74). Additionally, the levels of cytotoxic T Lymphocyte antigen 4 (CTLA-4) are higher in CSCs than those in normal cancer cells (75). CTLA-4, which functions as an analog of CD28, impedes T cell activation by hijacking CD80/86. Moreover, CTLA-4 presence in CSCs enhances the expansion of Tregs, which primarily suppress the anti-tumor T cell response (76). Furthermore, CSCs avoid being killed by cytotoxic CD8+ T cells by downregulating the expression of MHC I molecules (77).
Redox signaling determines the fate of T cell
Cellular redox reactions are tightly regulated by GSH dynamics and are essential for various cellular functions. Among the products of redox reactions, ROS are associated with a broad spectrum of T cell functions and fate (78). There are two major sources of ROS in T cells. Initially, NADPH oxidase (NOX) is activated by TCR stimulation, leading to the accumulation of cytosolic ROS in T cells (79). Subsequently, T cell generate adenosine triphosphate (ATP) to support the metabolic demand for their survival and function. Mitochondria are essential for ATP production. It drives multiple oxidative reactions of the TCA cycle products, NADH and FADH2, which activate the electron transport chain for a proton gradient across the mitochondrial membrane. This gap facilitates proton flow to activate ATP synthase, which generates ATP (79). During this process, the antioxidant system is activated to modulate redox homeostasis. Normally, antioxidant systems comprise antioxidant enzymes, such as SODs, catalases, glutaredoxins, sulfiredoxins, GPXs, peroxiredoxins, TRXs, thioredoxin reductases, methionine sulfoxide reductases, GSRs, and non-enzymatic molecules, such as pyruvate, GSH, ascorbate, oxaloacetate, and alpha-ketoglutarate (80).
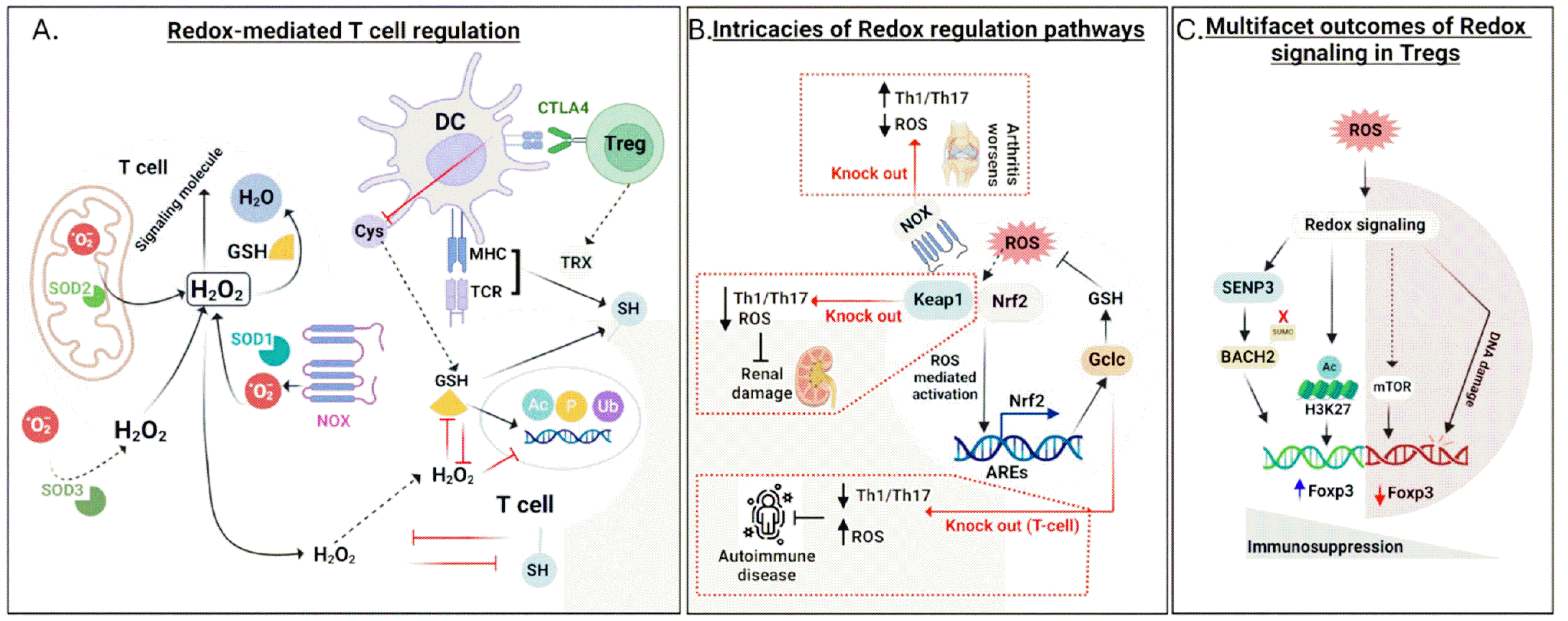
There are multiple mechanisms by which ROS regulate T cell fate. H2O2, a member of the ROS family, can bind to cysteine residues of signaling molecules to mediate post-translational modifications through its oxidizing effects (Fig. 5A) (81). Because excessive oxidation by ROS results in damage to T cells via the disruption of DNA and proteins, antioxidant systems should act immediately after ROS generation. As mentioned previously, GSH plays an integral role in ROS buffering. Given that cysteine and its thiol groups are the principal components of GSH, the immune system has evolved to utilize them for redox homeostasis. Cysteine secreted by DCs is taken up by T cells, which increase GSH levels (82). GSH maintains cell surface thiols in a reduced state and titrates intracellular H2O2 to a level that modulates DNA synthesis and epigenetic control (83). Moreover, T cells, DCs, and Tregs secrete another type of antioxidant, TRX, which preserves the reduced form of thiols on the cell surface (84). Tregs suppress the release of cysteine from DCs in a CTLA4-dependent manner, which in turn decreases the GSH levels in T cells (Fig. 5A) (85). This reduced GSH level permits ROS accumulation, thereby exerting a detrimental effect on T cell activation.
Diverse preclinical experiments have been performed using genetic and chemical interventions to explore the significance of redox signaling in disease settings. The genetic ablation of genes that modulate ROS generation has provided insights into their roles in the fate of T cells. In a mouse model of arthritis, NOX deletion reduced cytosolic ROS levels in CD4+ T cells. This outcome causes the expansion of Th1 and Th17 cells while decreasing Treg abundance, which exacerbates arthritis severity (Fig. 5B) (86). This result demonstrates that ROS signaling maintains immune homeostasis by balancing the ratio of inflammatory T cells to Tregs.
Another study focused on the function of the Nrf2 negative regulator Keap1. Upon Keap1 deletion in mice, cellular ROS levels dramatically decrease owing to the enhanced antioxidant capacity of Nrf2 (Fig. 5A) (87). Concomitantly, this leads to the expansion of Tregs and reduction of Th1 and Th17 cells, conferring protection against ischemia-reperfusion-induced acute kidney injury (Fig. 5A) (87). This tendency has been confirmed in a glucose-aggravated autoimmune condition as well (88). High glucose supplementation preferentially converts CD4+ T cells into autoimmune Th17 cells, displaying elevated levels of mtROS. By reversing mtROS via the antioxidant MitoQ, Th17 cells return to the level in healthy animals, which contributes to the mitigation of autoimmune pathology (88). Collectively, these results attempted to modulate ROS generation to investigate how the redox status affects the fate of T cells; however, because opposite consequences were observed at low ROS levels, the issue remains unclear.
The complex role of ROS in T cells has been elucidated under GSH-deficient conditions (89). The conditional deletion of GCLC prevents GSH production. In the absence of GCLC, T cells showed normal activation at the early stage of TCR stimulation but were unable to become intact effector cells. This resulted from a defect in the Myc-mediated metabolic process in the absence of GSH, which triggered autoimmune resistance in mice by reducing the number of Th1 and Th17 cells (Fig. 5B). This study suggests that T cells are extremely sensitive to ROS levels, which presumably supports the understanding of the controversies reviewed in an earlier section.
Tregs control immune homeostasis by restricting aberrant inflammation; however, in the TME, they support tumor growth. The nature of Tregs is governed by ROS signaling. The complex network of Tregs is critical for maintaining Tregs, and the TF BACH2 is indispensable for Treg stability, which results from the BACH2-mediated repression of the effector T cell gene signature (90). ROS mediate SUMOylation of BACH2 and supports the suppressive activity of Tregs (91). SUMOylation is a post-translational modification that depends on the extent to which SUMO binds to target proteins. This process is coordinated by several enzymes that add or remove SUMO from proteins and the stability and localization of TFs are determined by SUMOylation (92, 93). Conditional deletion of the deSUMOylating enzyme SUMO-specific protease 3 (SENP3), in CD4+ T cells disrupted immune homeostasis in mice, expanding the inflammatory population of T cells (91). However, CD4+ T cells exhibited activated phenotypes in response to in vitro TCR stimulation regardless of SENP3, but SENP3 deficiency in Foxp3-expressing cells reduced the quantity of Tregs. This indicates that SENP3 preserves immune homeostasis by promoting Treg maintenance. Mechanistically, SENP3 deSUMOylated BACH2, rendering BACH2 remained in the nucleus (Fig. 5C). This localization enhances the stability of Foxp3 and represses the transcriptional network of inflammation, promoting the Treg-mediated exacerbation of tumor growth. During this process, ROS induced SENP3 accumulation, indicating that the redox status affects SUMOylation in Tregs (Fig. 5C). Recently, epigenetic regulation of Tregs by ROS was identified in the context of bile acid-mediated Treg induction (94). During Treg differentiation, one of the bile acid metabolites, iso-alloLCA, increased mtROS levels in CD4+ T cells and enhanced the expansion of Tregs (Fig. 5C). mtROS caused elevated levels of H3K27 acetylation at the Fopx3 promoter (Fig. 5C), accelerating the transcription of Foxp3.
As observed in T cells, ROS also play a contradictory role in Tregs. Under autoimmune conditions, Tregs fail to adapt to altered mitochondrial metabolism, which leads to mtROS accumulation and decreased DNA stability (95). DNA damage in Tregs causes cell death and disrupts Treg-mediated immune homeostasis (Fig. 5C). Collectively, targeting ROS signaling is expected to pave the way for the development of T cell-modulating therapeutics; however, further investigation of the spatiotemporal functions of ROS in T cells is needed.
Future Perspectives and Conclusions
GSH dynamics in the TME critically affect the fate of CSCs and T cells. In CSCs, oxidative stress induces NRF2-dependent activation of the GSH antioxidant system, which may promote differentiation into bulk tumor cells (Fig. 3B, 3C) (46). The mtGSH dynamics in CSCs might regulate their stemness via scavenging mtROS derived from SOD2K68Ac which activate HIF-2α-dependent pluripotent TFs (Fig. 1, 3B) (34). Membrane GSH protects against membrane lipid peroxidation and ferroptosis and enhances metastasis (43, 44, 57). Simultaneously, the pro-tumoral function of T cells in the TME presents a significant barrier to achieving effective antitumor activity. Deleting these T cells has been effective in preclinical tests for cancer therapy, but its efficacy and safety should be proven to treat cancer patients in the clinic. Targeting GSH dynamics may become another option to modulate T cells in the TME, based on the compelling evidence suggested here. The function of Tregs/Th17 cells is tightly regulated by multiple mechanisms of the REDOX reaction, and these T cells aggravate tumor severity by communicating with CSCs via physical contact and cytokines. Because CSCs are also regulated by GSH levels, exploring the GSH code would provide clues for deciphering the network of CSCs and T cells in the TME. Therefore, the GSH dynamics in the TME may be a potential target for tumor therapy. However, the GSH antioxidant system is replaced by the TRX and DUB pathways in CSCs (Fig. 3B). The combinatorial targeting of both the GSH system and certain compensation pathways results in synthetic lethality (46, 52, 53, 59, 96, 97), suggesting that cellular antioxidant systems are good candidates for combination therapy with other cancer drugs (32, 98). To develop efficient strategies for tumor treatment, accurate tools for the temporal and spatial analysis of GSH dynamics within CSCs and T cells in the TME are required. Recent advances such as real-time live-cell GSH monitoring probes may be helpful (12-15).
 XML Download
XML Download