

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Head traumas are among the important health problems that may result in death or disability and require long-term treatment and care. Social and technological life, which is accelerating day by day, increases the incidence, mortality and morbidity risk of head traumas [7,36]. The World Health Organisation has reported that 3.5 million people worldwide die annually as a result of trauma [4]. According to USA data, 1.4 million people are treated for traumatic brain injury (TBI) and 50 thousand patients die annually [17]. The group with the highest risk is individuals aged 15–24 years. The incidence rate in males is three times higher than in females [18,22]. Deaths due to head trauma constitute 2–4% of all deaths and more than half of trauma-related deaths [8,32]. The mortality rate in head trauma is highest in the early period following trauma [27]. Half of the head traumas with a high risk of death result in death at the scene or in emergency transport vehicles. Two third of patients with head trauma with a high risk of death who can be brought to hospital and examined by a physician die within the first 24 hours [21].
In head trauma, the injury that occurs with vascular or neuronal injury as a result of direct mechanical effects of the trauma force is called primary injury. The development of space-occupying events and damage such as ischaemia as a result of complications secondary to the primary injury is defined as secondary injury [27]. Local lesions that occur in a specific region after the direct effect of trauma are examples of primary injury, while the sequence of pathophysiological events triggered in the brain by diffuse or local lesions and usually ischaemia are examples of secondary injury [2,13].
Primary brain injury includes scalp injuries, skull fractures, parenchymal contusion, intracranial hemorrhage such as subdural hemorrhage, epidural hemorrhage, intracerebral hemorrhage and lacerations. Secondary injury may occur hours or days after the primary injury. It occurs due to physiological and pathological events triggered by the primary injury. Secondary injury is a poor prognostic indicator in TBI. Free radicals, mitochondrial dysfunction and inflammation are the mechanisms involved in secondary injury. In addition, excitatory receptor activation, ischaemia, inadequacy in membrane ion pumps, increase in arachidonic acid metabolism and acute brain oedema also cause secondary injury [16].
Lipid peroxidation is responsible for the production of reactive oxygen species (ROS) released after trauma, which is an important cause of secondary brain injury [34]. Therefore, in order to prevent neuronal damage and improve neurological outcomes, it is necessary to prevent ROS formation or distribution to cells. Agents that inhibit ROS have been reported to have a positive effect on recovery after events such as ischemia and trauma that worsen the neurological picture [30]. Antioxidants such as catalase (CAT), glutathione peroxidase (GPx), superoxide dismutase (SOD) have been reported as ROS inhibitors [10].
The aim of this study was to investigate the possible neuroprotective effects of dexpanthenol (DXP), which has been proven to be neuroprotective in secondary mechanisms (such as ischemia-reperfusion, asthma) [14] that play a role in the pathophysiology of secondary injury after head trauma, such as impaired blood brain barrier permeability, increased oxidant damage, development of cerebral oedema and other mechanisms leading to brain injury, in an experimental trauma model.
MATERIALS AND METHODS
The study protocol was approved by the Ethics Committee on Animal Research at the Faculty of Medicine, Inonu University, Malatya, Türkiye (ethical approval No. 2020/01-3).
Research animals
Thirty-six female Wistar-Albino rats, 6 months old, weighing between 220–285 g, which had not been used in any previous experiment, were included in the study. All rats were reared in polycarbonate cages with four rats in each cage. Ambient temperature and humidity (22°C±3°C and 60%±7% humidity) were kept within a constant range. All cages were cleaned daily. All rats were fed with standard animal feed and sufficient water. Water was given in stainless steel feeding bottles. Food was given in steel cages. Throughout the experiment, the environment was illuminated with 12-hour light-dark cycles at appropriate wavelengths.
Rats were divided into four main groups. Blood samples were taken from all rats 72 hours after head trauma for biochemical examination and cerebral tissue samples were taken for immuno-histochemical and histopathological examination after decapitation under general anaesthesia. 1) Group 1 (control group; n=8), no head trauma was induced in rats in this group and no treatment was given; 2) group 2 (trauma group; n=10), rats in this group were subjected to head trauma and no treatment was given; 3) group 3 (trauma + DXP group; n=10), rats in this group were subjected to head trauma. DXP was administered intraperitoneally (i.p.) at a dose of 500 mg/kg six times at 30 minutes, 6, 12, 24, 36, and 48 hours after trauma; and 4) group 4 (DXP group; n=8), DXP was administered i.p. simultaneously with the rats in the trauma + DXP group without causing head trauma.
A pilot study was performed on two old rats, which we did not include in the study. The reason for the pilot study was the high mortality rate immediately after trauma. Based on these reasons, the height was reduced to 50 cm and the weight to 350 g in the present study. In addition, the head protective material in the parietal region was removed for modelling the previously planned sudden free falls and motor vehicle injuries without helmet. For this reason, the method used in trauma was named as ‘modified Marmarou weight-reduction model’ [20]. With this modified method, the mortality rate after TBI was reduced to zero.
Preparation of the rats
Xysilazine hydrochloride (HCL) 5–10 mg/kg i.p. and ketamine 50 mg/kg i.p. were administered to rats to induce anaesthesia. After administration of pharmacological agents, rats were observed in their cages until deep anaesthesia occurred. The rats were then removed from their cages and weighed with a precision balance and their weights were noted. The rats were placed on the tensioned foil in prone position. The skull was placed 50 cm below the bottom hole of the open-ended tube containing the weight. The mass weighing 350 g was placed on the upper end of the tube in a free fall. The 350 g mass was released in a free fall to cause head trauma. A single trauma was induced in all rats.
Creation of head trauma
Head trauma was induced by modifying the free fall model described by Marmarou et al. [19]. The rats were fasted 24 hours before the operation by giving only water. Xylazine HCL 8 mg/kg and ketamine HCL 75 mg/kg were administered i.p. as anaesthetic agents. Anaesthetised rats were placed in supine position. After trauma, the incision site was sutured closed and the rats were released for food and fluid needs.
Collection of samples
Deep anaesthesia was induced in all rats using ketamine HCL and xylazine after a predetermined period of 72 hours post-trauma. Following deep anaesthesia, the brain tissue was removed without damage. The brain was divided into two hemispheres and the hemispheres were placed in previously prepared and numbered containers for each subject. Brain samples were placed in 10% formol solution for histological examination and kept at -70°C for biochemical examination.
Preparation of homogenates
Each 0.1 g of brain tissue was reacted with 0.9 mL of sodium phosphate containing protease inhibitor (Bishop) at pH : 7 and 100 mM. Ice was added to the product. Centrifuged at 14000 rpm for 3 minutes and 10% tissue homogenates were prepared. These homogenates were centrifuged at 3500 rpm for 15 minutes at +4°C and supernatants were obtained. Supernatants and serum samples were used for malondialdehyde (MDA), SOD, GPx, CAT, glutathione (GSH), tissue protein measurements and histological examinations.
Measurement of tissue thiobarbituric acid reactive products levels and MDA levels
MDA levels were determined according to the method proposed by Ohkawa et al. [23]. A pink coloured chromogen was formed by the heating reaction of MDA with thiobarbuturic acid in acid medium. The amount of MDA was determined as nmol/g wet tissue by measuring the intensity of pink colour at 532 nm wavelength in spectrophotometer (Microplate reader; BioTek Synergy H1, Winooski, VT, USA). The intensity of the pink colour is directly proportional to the MDA concentration in the tissue.
Tissue SOD activity measurement
SOD enzyme activity levels of tissue samples were measured according to the method proposed by Sun et al. [31]. SOD enzyme activity is measured by the reduction of superoxide radicals by nitroblue tetrazolium (NBT) to produce colour in the medium. Superoxide radicals produced by xanthine-xanthine oxidase system are used in the reduction of NBT. It forms blue coloured formazone with the reduction reaction giving maximum absorbance at 560 nm. This colour was measured in spectrophotometer and enzyme activity was found. SOD enzyme reduces the reduction reaction by decreasing the superoxide anion in the environment. The blue colour formed is in light tones. When there is no enzyme in the medium, the reduction reaction is maximum and a dark blue colour is formed. The amount of SOD in the tissue and the intensity of blue coloured formazone are inversely proportional. SOD activity was determined by detecting the change in absorbance at 560 nm wavelength in spectrophotometer (Microplate reader; BioTek Synergy H1). Tissue SOD units were given in U/g protein.
Tissue Gpx activity measurement
Gpx activity was performed according to the method proposed by Paglia and Valentine [25]. Gpx is an enzyme that catalyses the reaction of hydrogen peroxide (H2O2) with reduced glutathione. The products of the reaction are H2O2 and oxidised glutathione. With this reaction, the radical H2O2 is converted to water. Reduced glutathione, the substrate of this reaction catalysed by Gpx, reacts with nicotinamide adenine dinucleotide phosphate (NADPH) and is produced in the presence of GSH reductase enzyme. NADPH must be used continuously to remove H2O2 from the medium. NADPH, which shows maximum absorbance at 340 nm wavelength in spectrophotometer, decreases with the presence of Gpx. By measuring the decrease in NADPH absorbance in spectrophotometry (Microplate reader; BioTek Synergy H1), Gpx level in the tissue was expressed as U/g protein. The increase in Gpx activity in tissue is proportional to the negative change in NADP absorbance.
Measurement of tissue CAT enzyme activity
The CAT activity of tissue samples was measured by the method proposed by Aebi [1]. H2O2 produces a maximum absorbance at 240 nm in a spectrophotometer. CAT reacts with H2O2 and breaks it down into water and oxygen. CAT enzyme activity was measured by decreasing the absorbance of H2O2 in a spectrophotometer (Microplate reader; BioTek Synergy H1). The unit is K/g protein. The decrease in absorbance is directly proportional to the CAT enzyme activity in the medium.
Tissue reduced GSH measurement
GSH levels in tissue samples were measured by the method proposed by Ellman [5]. According to Ellman’s method, GSH and 5,5’-dithiobis-2-nitrobenzoic acid react. The reaction produces a yellow-green colour in the medium. The intensity of the yellow-greenish colour was measured in a spectrophotometer (Microplate reader; BioTek Synergy H1) at 410 nm wavelength and the amount of reduced GSH was found in nmol/g wet tissue. The intensity of yellow-greenish colour is directly proportional to the GSH concentration in the medium.
Tissue protein measurement
Protein levels in tissues were measured by the method proposed by Lowry et al. [15]. pH : 10–10.5, the peptide nitrogen of the protein reacts with copper (Cu+2) ions to form the protein-copper complex. Phosphomolybdate-phosphotungstate reagent (Folin-Ciocalteu-Phenol reagent) was added to the protein-copper complex. The reaction of the protein-copper complex with phosphomolybdate-phosphotungstate reagent produces blue-coloured heteromolybdenium. The aromatic amino acids tyrosine and tryptophan in proteins are the reducing structures that produce the blue colour. Therefore, the intensity of the colour is directly proportional to the amount of protein in the medium. The unit of protein measurement is mg/mL. This method can detect even protein concentrations of 0.1 mg/mL.
Histological examination
At the end of the study, brain tissue samples of decapitated rats were taken for histological evaluations. The tissue samples taken for histopathological examinations were fixed in 10% formaldehyde and embedded in paraffin blocks. Five µm thick sections cut from the blocks were stained with Haematoxylin and Eosin stain. Light microscopy was used for histological evaluation. The sections were evaluated for congestion, cell infiltration, vascular congestion, hemorrhage and neuronal degeneration in the piamater layer. Microscopic damage was determined as none (0), mild (1), moderate (2), and severe (3) for each criterion. The sections were examined and photographed using ‘Leica DFC 280’ brand light microscope and ‘Leica Q Win Image Analysis System’.
Statistics
IBM SPSS for Windows version 26.0 software (IBM, New York, NY, USA) was used for all statistical analyses. Data were presented as mean±standard deviation and median (minimum-maximum) for continuous variables. Normality distribution of the data was analysed by Shapiro Wilks test. Mann Whitney U test was applied for pairwise independent group comparisons according to normality distribution characteristics.
RESULTS
Histological results
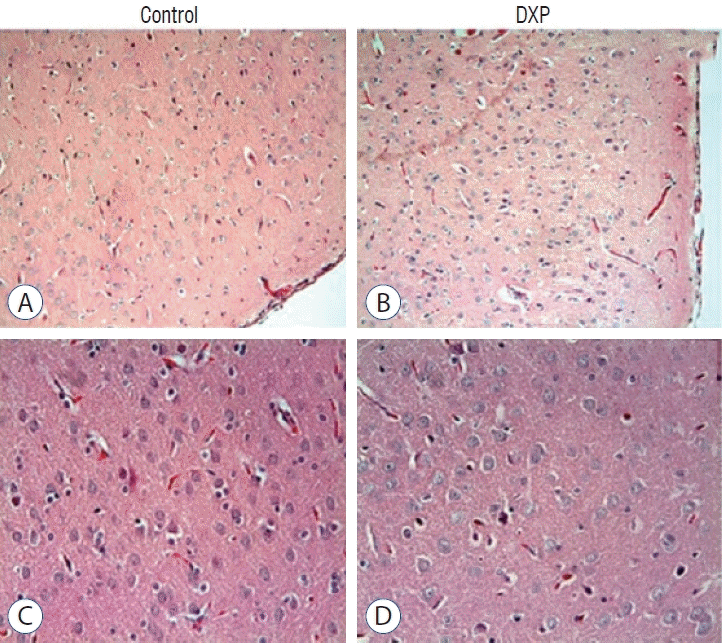
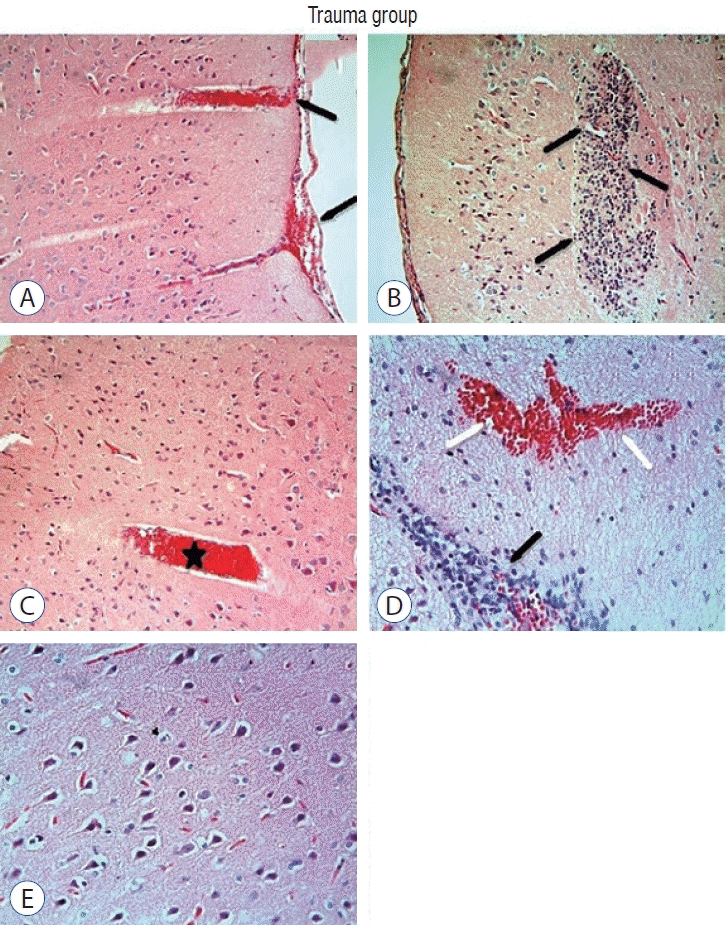
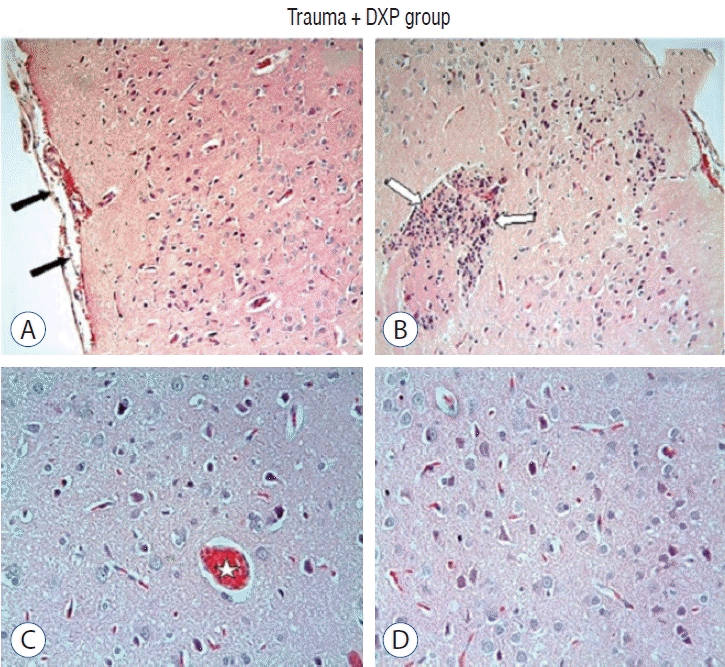
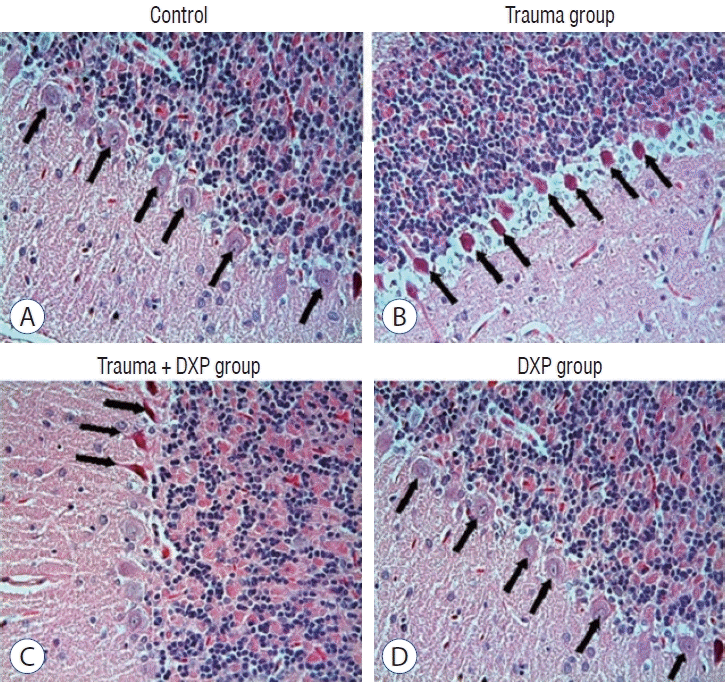
Light microscopic examination of brain tissue samples of rats in group 1 (Fig. 1A and C) and group 4 (Fig. 1B and D) showed normal histological appearance. In the brain tissue of rats in group 2, congestion in the piamater layer (Fig. 2A), cell infiltration (Fig. 2B and D), vascular congestion (Fig. 2C), hemorrhage (Fig. 2D), and neuronal degeneration (Fig. 2E) were observed. In group 3 (Fig. 3), a decrease in histopathological damage in brain tissue was observed. In this group, a small amount of congestion in the piamater layer (Fig. 3A), cell infiltration (Fig. 3B), vascular congestion (Fig. 3C), and neuronal degeneration (Fig. 3D) were significantly reduced. In group 1 (Fig. 4A) and group 4 (Fig. 4D), cerebellum tissue and Purkinje cells had normal histological appearance. In group 2 (Fig. 4B), many degenerated Purkinje cells were observed. In group 3 (Fig. 4C), degenerated Purkinje cells were significantly reduced.
When the histopathological scores between the groups were compared, it was observed that the histopathological score was significantly higher in the trauma group compared to the control, trauma + DXP and DXP groups, and the histopathological score was significantly higher in the trauma + DXP group compared to the control and DXP groups (Table 1).
Biochemical results
MDA and GSH levels in samples taken from the brain tissue of rats in the trauma group (group 2) were significantly higher than in the control group (group 1) (p<0.05) (Table 2). MDA levels in samples taken from the brain tissue of rats in the control group (group 1) were significantly higher than in the DXP group (group 4) (p<0.05) (Table 2). When MDA, GSH, SOD, CAT, and GPx levels were compared in samples taken from the brain tissue of rats in trauma + DXP (group 3) and trauma (group 2) groups, it was observed that MDA and GSH levels were significantly lower and SOD, CAT, and GPx levels were significantly higher in samples taken from the brain tissue of rats in group 3, which received DXP after trauma, compared to group 2 (p<0.05) (Table 2).
MDA levels in serum samples of rats in the control (group 1) group were significantly lower than those in the trauma (group 2) group (p<0.05) (Table 3). GSH and SOD levels in serum samples of rats in the control (group 1) group were significantly lower than those in the DXP (group 4) group (p<0.05) (Table 3). There was no significant difference in the levels of MDA, GSH, SOD, CAT, and GPx in the serum samples of rats in the DXP group (group 3) and trauma group (group 2) (p>0.05) (Table 3).
DISCUSSION
TBI is an injury with high morbidity and mortality rates worldwide. This makes it a global health problem [2,4,7,13,27,28,36]. In order to minimise cerebral injury after TBI, numerous studies have been conducted to prevent the initial impact injury and to seek treatment to prevent subsequent neuronal damage, to increase neuronal reorganisation and functional recovery [12,26,29]. Currently, there is no effective treatment for first impact injury. In developed countries, interventions by public health organisations and experts, such as seat belt legislation, helmet use and workplace health and safety regulations, have helped to reduce the incidence of severe TBI. Experimental studies on the pathophysiology of TBI with neuroprotective effects have not been successfully adapted to clinical treatments. Nevertheless, promising efforts are underway to optimise intensive care protocols, develop an evidence base for surgical intervention and translate promising neuroprotective pharmacotherapies into application to human pathology [35]. The large number of animal models available, the continuous description of new methods and the lack of standardisation show that the literature cannot agree on a common consensus on the head trauma model. Therefore, although our study uses a model supported by the literature, it may have deficiencies due to the above reasons. All these reasons may affect the clinical response of the efficacy of the drug we expect [3,35]. Marmarou’s weight reduction model is a standard model for experimental treatments after TBI observed in patients with head trauma and is the most frequently used trauma model to create experimental TBI. Although it is not possible to reproduce all pathophysiological changes of diffuse axonal injury (DAI) with this method, it is possible to reproduce biomechanical changes and clinical features after trauma. It also provides the development of brain oedema between 6–24 hours after head trauma [31]. Because of these effects, this model was used in our study.
Recent studies have been mainly aimed at preventing secondary pathophysiological events after diffuse TBI and developing medical treatment, and promising results have been obtained from these studies [12,26,29]. Based on these promising studies, we aimed to investigate the neuroprotective efficacy of DXP, which was previously known to have anti-inflammatory and anti-cytotoxic properties.
In our study, MDA and GSH levels were found to be low and SOD, CAT, and GPx levels were found to be significantly higher in the dexpentanol-treated group after trauma. The similarity of the biochemical parameters and the concordance of the results showed that dexpentanol decreased MDA level and increased SOD, CAT, and GPx levels in support of lipid peroxidation and inflammatory response. In a recent study by Toplu et al. [33], DXP was tested in the treatment of cisplatin-induced autotoxicity. Biochemical analyses showed an increase in MDA, TOS, and OSI in the cisplatin group compared to the control group. SOD, CAT, GPx, and TAS parameters were decreased in the cisplatin group compared to the control group. An increase was detected in the treatment group given DXP.
Brain injury in TBI includes primary injury caused by direct or indirect contusion, subdural haematoma and cerebral ischaemia resulting in shearing or stretching of brain tissue, secondary injury characterised by DAI and inflammatory reactions, and regeneration. The secondary, non-mechanical injury stage is progressive and lasts from hours to days. For these reasons, the drug to be developed in treatment should not be given as a single dose. In this context, in our study, the drug was administered regularly at 30 minutes, 6, 12, 24, 36, and 48 hours to prevent progressive damage continuously.
DXP has been reported to protect tissues against oxidative damage in various ischaemia/reperfusion models. Gülmez et al. [9] showed antioxidant, anti-inflammatory and anti-apoptotic effects of DXP in neurons as a result of biochemical, histopathological and neurological examinations in experimental rabbit spinal cord ischemia/reperfusion injury (SCIRI) model. Lower levels of malondialdehyde and caspase-3 were detected in neurons with DXP treatment after SCIRI in rabbit spinal cord. While myeloperoxidase and xanthine oxidase activities decreased, CAT activity increased. Although ischaemia/reperfusion injury does not cause direct mechanical trauma, it leads to secondary TBI due to metabolic events and axonal conduction change [9]. Apart from ischaemia/reperfusion injury, mechanical tissue damage caused by direct trauma and pathophysiological events that develop after neuronal loss were thought to affect the neuroprotective properties of DXP.
Karahan et al. [11] investigated the effect of DXP on nerve healing following neurorrhaphy in ruptured peripheral nerves and found significantly lower MDA levels. DXP has been shown to play a positive role in peripheral nerve healing by reducing lipid peroxidation. Better histological results were obtained with less fibrosis during healing. It has been shown that the compound muscle action potential is higher and the action potential delay is lower in muscles stimulated by peripheral nerves treated with DXP after trauma. Thus, DXP, which has been shown to have a neuroprotective effect after TBI, has also been shown to have a positive effect on the recovery of the physiological function of neurons.
When the literature is examined, it is seen that there is a limited number of studies showing that DXP may have antiinflammatory and neuroprotective effects [6,11,12,24,26,37]. This situation reveals the originality of our study. At the same time, it is thought that the results obtained from our study will be a guide for future studies on the neuroprotective effect of DXP.
CONCLUSION
In conclusion, we think that DXP may be a useful option in the treatment of secondary brain injury after head trauma. In future studies on the neuroprotective efficacy of DXP in head trauma, it would be useful to use larger experimental groups and different dose treatment schemes. We hope that DXP, whose neuroprotective efficacy has been shown in experimental models, will also be shown to be effective in clinical studies.
 XML Download
XML Download