

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Chronic rhinosinusitis (CRS) is defined as inflammation of the mucosa of the nose and paranasal sinuses that persists for more than 12 weeks [1]. Typical symptoms include facial pain or pressure, nasal discharge, congestion, hyposmia, and anosmia. Patients with CRS often have coexisting inflammatory airway conditions, such as asthma or allergic rhinitis.
CRS can be categorized into different phenotypes based on clinical findings, immunological status, and genetic diversity. The basic classification relates to the presence of nasal polyps, resulting in two main types: CRS without nasal polyps (CRSs-NP) and CRS with nasal polyps (CRSwNP). Each phenotype can be further divided into endotypes (types 1, 2, and 3) based on inflammatory signatures reflecting patterns of structural remodeling and inflammation.
The inflammatory signature determines the CRS endotype, influencing disease severity, comorbidities, prognosis, and response to treatment. CRS is a highly prevalent disease, affecting 10.9% of the population in Europe based on EPOS (European Position Paper on Rhinosinusitis and Nasal Polyps) criteria. In China, the prevalence stands at 8%, in Korea at 11%, and in the USA, it varies between 4.8% and 12% [2].
Notable geographic variations have been reported in the prevalence of type 2 and non-type 2 dominant inflammation in the sinonasal mucosa of CRS patients. In Western countries, up to 80% of CRSwNP cases are identified as type 2 responses, characterized by eosinophilic infiltration and elevated interleukin (IL)-5 levels. In contrast, CRSwNP in Chinese patients predominantly features type 1 and type 3 inflammatory profiles. Additionally, 33.3% of nasal polyps in Korean patients are eosinophilic [2].
Recent longitudinal studies have shown a significant rise in type 2 inflammatory CRSwNP in Asia over the past decade, which is suspected to be linked to nasal colonization by Staphylococcus aureus [1].
Our understanding of the pathophysiology of CRS has evolved from a model focused primarily on the physical obstruction of sinus cavity ventilation and drainage. It now embraces a mucosal concept that underscores the complexity of mucosal immunologic variations and cellular aberrations. This modern perspective emphasizes the epithelial barrier’s response to external insults, including pathogens, allergens, and environmental toxins [3].
Damage to the epithelial barrier initiates a repair process that reactivates the epithelial-mesenchymal transition (EMT) [4]. In the EMT, epithelial cells lose their characteristics and gain mesenchymal properties, facilitating tissue repair and remodeling. The EMT plays a significant role in the pathogenesis of CRS by contributing to tissue fibrosis, thereby perpetuating the disease state. The reactivation of the EMT in response to epithelial damage underscores the importance of maintaining epithelial integrity in managing CRS and preventing its progression.
This review focuses on the EMT in CRS, exploring how disruptions to epithelial cell integrity and subsequent EMT processes make a major contribution to the pathogenesis and progression of CRS, thereby offering potential therapeutic approaches.
EPITHELIAL-MESENCHYMAL TRANSITION
The EMT involves complex changes in the cell phenotype, characterized by the loss of apical-basal polarity in epithelial cells, cytoskeletal reorganization, and diminished cell-cell adhesion. During this process, cells either individually or collectively adopt mesenchymal traits, which increase their motility and invasive potential. Typically, post-EMT, cells transition from using cytokeratin to vimentin for intermediate filaments, and cortical actin filaments are substantially rearranged.
Cells can exist anywhere along the epithelial-mesenchymal spectrum, a concept referred to as “epithelial-mesenchymal plasticity.” In most cases, cells exhibit a combination of epithelial features (e.g., cytokeratin) and mesenchymal traits (e.g., cell motility) along with the corresponding markers.
The numerous molecules involved in the EMT can be categorized into three groups: EMT-inducing signals, EMT transcription factors (EMT-TFs), and EMT markers. These molecules define and constitute various epithelial and mesenchymal cell characteristics [5].
Key transcription factors such as the Snail, Slug, Twist, zinc finger E-box binding homeobox (ZEB) 1/2, and paired related homeobox (PRRX) families play central roles in controlling cell adhesion, migration, and extracellular matrix (ECM) degradation. These factors are conserved across various biological contexts in executing the EMT [5]. The loss of epithelial properties is mainly driven by the Snail and ZEB families, which also interact with the main mesenchymal inducers Twist and PRRX1 [6].
Tight junction disintegration is an initial step in the EMT, leading to the repositioning of occludin, claudin, and zonula occludens (ZO)-1, as well as the loss of cell polarity and changes in the cytoskeletal structure and ECM. As cell adhesion weakens, E-cadherin—a crucial EMT marker—is lost, which acts as a fundamental trigger for the EMT. This disintegration of epithelial junctions allows cells to take on mesenchymal traits, resulting in a notable increase in the expression of Ncadherin, vimentin, α-smooth muscle actin (α-SMA), collagen, fibronectin, and other extracellular matrix proteins. The activity and expression of extracellular proteases such as matrix metalloproteinase (MMP)-9 also rise, promoting the breakdown of ECM components [7].
The regulation of the EMT is complex, ensuring the preservation of the epithelial phenotype while allowing for transient plasticity to maintain homeostasis during wound repair. Additionally, EMT activation increases plasticity in adults under various pathological conditions, including cancer metastasis and chronic inflammation. In inflammatory contexts, EMT can drive tissue degeneration, leading to severe diseases such as fibrosis [6] .
EPITHELIAL-MESENCHYMAL TRANSITION IN CHRONIC RHINOSINUSITIS
The EMT has been observed in both phenotypes of CRS, with mesenchymal markers being overexpressed and correlating with disease severity and tissue inflammation [8].
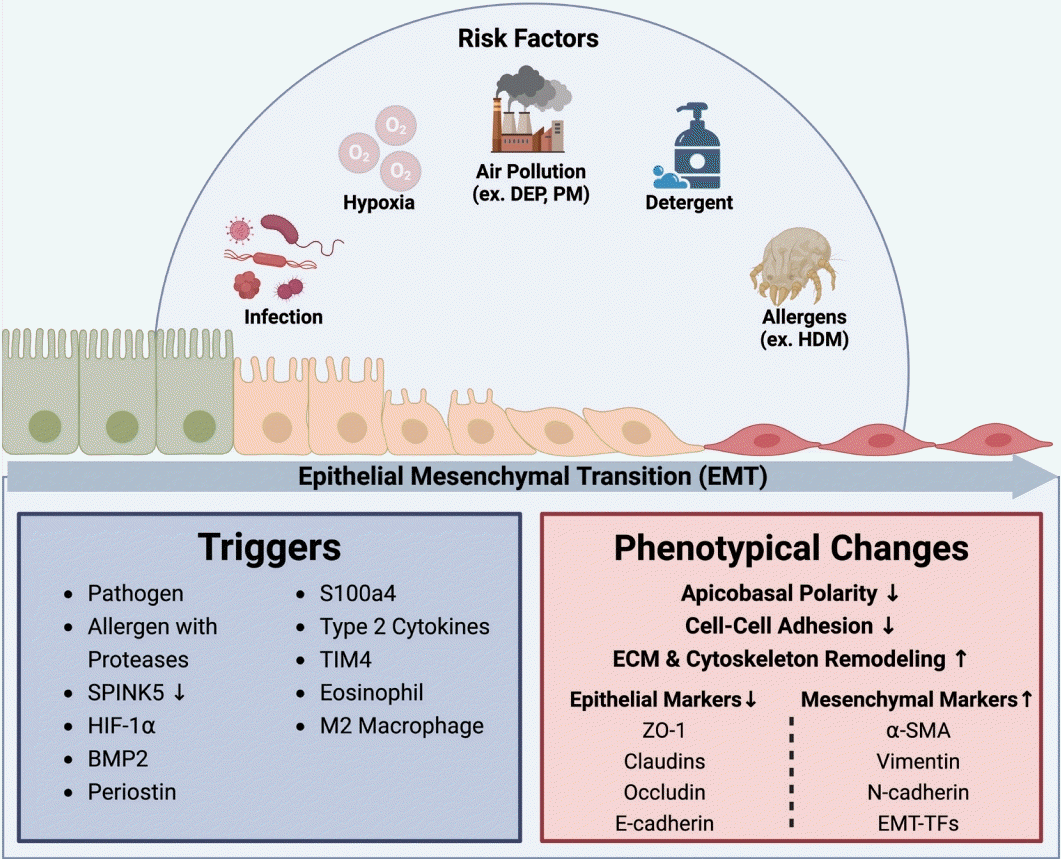
In CRS, the EMT can be triggered by various factors, including respiratory pathogens, allergens with proteases, reduced levels of the protease inhibitor serine peptidase inhibitor Kazal type 5 (SPINK5), levels of hypoxia inducible factor (HIF)-1α, smoking, bone morphogenetic protein 2 (BMP2), periostin, lipopolysaccharide, and the type 2 cytokines IL-13 and IL-4 (Fig. 1) [1,4,9,10]. These triggers activate several molecular pathways and signaling molecules, such as transforming growth factor (TGF)-β, members of the epidermal growth factor (EGF) family, vascular endothelial growth factor, fibroblast growth factor, tyrosine kinase pathways, Notch, proconvertase 1, SMAD pathways, Wnt, signal transducer and activator of transcription 3 (STAT3), ganglioside GD3, and gp130–Src–YAP [1,4,11].
These changes result in alterations of the function of transcription factors, such as Snail, Slug, Twist, and ZEB1/2. These transcription factors orchestrate the complex gene expression changes driving the EMT process in CRS. Notably, EMTTF markers exhibit different patterns depending on the patient and their anatomical location. For instance, Snail and Slug levels in the nasal polyps of CRSwNP patients are significantly higher than in patients with healthy inferior turbinates [12]. Conversely, Twist levels are elevated in nasal epithelial cells (NEC) of the inferior turbinate, whereas levels of Snail and N-cadherin increase in nasal polyps [12]. These observations suggest that these markers are differentially regulated in CRS lesions. Twist levels are higher in CRSwNP patients than in healthy individuals, with its overexpression downregulating epithelial gene expression and activating mesenchymal gene expression [12]. ZEB1 expression is significantly higher in CRSwNP patients, correlating with severe disease symptoms and inflammatory responses. In human nasal epithelial cells, ZEB1 knockdown reduces EMT markers and pro-inflammatory factors, while overexpression increases them, suggesting that ZEB1 facilitates the EMT and promotes inflammation [13]. Baseline ZEB2 levels are increased in CRS patients and further upregulated in those with nasal polyps, while E-cadherin shows opposite trends. CRSwNP exhibits more severe EMT characteristics than CRSsNP and normal tissues, supporting the possibility that ZEB2 is upregulated in NP-derived epithelial cells [14].
In CRS, the expression levels of junctional proteins such as E-cadherin, claudin, occludin, and ZO-1 notably decrease. Similarly, the epithelial marker cytokeratin-18 shows reduced expression. Conversely, levels of EMT-associated markers, including periostin, laminin, vimentin, fibronectin, MMP2, MMP9, tenascin-C, α-SMA, fibroblast-specific protein 1, Snail, and Slug, significantly increase (Fig. 1) [4].
RISK FACTORS
Infection
Infections are significant risk factors for the development and exacerbation of CRS. Viral infections play a crucial role by inducing inflammation, maintaining persistent inflammatory stimulation during asymptomatic infections, and triggering acute symptom exacerbations. At the cellular level, respiratory viruses disrupt the barrier function of the sinus epithelium and weaken the robustness of the immune response following viral and bacterial infections [15]. Rhinovirus is known to induce the EMT. Furthermore, a synergistic effect was observed when human bronchial epithelial cell lines were co-treated with rhinovirus and TGF-β, further promoting the EMT process. Rhinovirus also disrupts barrier function in bronchial epithelial cells, exacerbating the pathological changes in CRS [16,17]. Persistent respiratory syncytial virus infection promotes the expression of oncogenes, cell proliferation, and EMT through nodal signaling pathways [18], highlighting the role of chronic viral infections in maintaining and worsening the disease state. Bacterial infections, such as those caused by Pseudomonas aeruginosa and S. aureus, can exacerbate the EMT. When THP-1 cells activated by P. aeruginosa are co-cultured with primary bronchial epithelial cells, they significantly promote the TGF-β1-induced EMT [19]. S. aureus also contributes to the EMT [20]. Its α-toxin disrupts cell morphology and integrity in human airway epithelial cells by impairing stable cell contacts and actin cytoskeleton organization [21]. Additionally, S. aureus and its products are implicated in the pathogenesis of nasal polyposis, with significant reductions in epithelial cell junction molecules such as E-cadherin, ZO-1, and occludin in mature ethmoidal polyps [22,23]. These interactions underscore the multifaceted impact of infections on the pathogenesis of CRS, particularly through mechanisms involving the EMT, and highlight the importance of managing both viral and bacterial infections to mitigate disease progression.
House dust mites and allergens
Exposure to house dust mites (HDMs) in the respiratory tract has been reported to cause significant airway inflammation and thickening of the smooth muscle layer in large airways. In mice, HDM exposure leads to increased levels of TGF-β1 in the airways, while epithelial cells lose the expression of E-cadherin and occludin and gain the expression of mesenchymal proteins such as vimentin, α-SMA, and procollagen I [24]. Furthermore the airway epithelial cells of mice exposed to HDMs showed increased expression of Snail1, which represses the transcription of E-cadherin and induces the EMT [24].
HDMs induce signaling through the EGF receptor (EGFR), increasing levels of vimentin and fibronectin and slightly decreasing E-cadherin levels due to EGF signaling. Inhibiting EGFR can prevent TGF-β/HDM-induced EMT. TGF-β was found to facilitate the dissociation of EGFR from E-cadherin and prolong EGFR signaling, promoting the EMT in TGF-β-primed epithelium exposed to HDM [25]. Additionally, HDM extracts increased the expression of IL-33 and CD146 in epithelial cells, contributing to airway remodeling and enhanced EMT [26]. HDM/TGF-β1 can also induce the EMT in human bronchial epithelial cells via the Sonic hedgehog signaling mechanism [27]. Protease 1 from Dermatophagoides pteronyssinus has been demonstrated to disrupt tight junctions by degrading key junctional proteins, such as ZO-1 and occludin [28]. Additionally, decreased levels of SPINK5 have been implicated in the loss of barrier integrity in the skin. Notably, reduced SPINK5 levels have also been observed in the nasal epithelium of patients with CRS, suggesting a potential mechanistic link between protease activity and epithelial barrier dysfunction in CRS [29,30]. These findings illustrate the substantial impact of HDM exposure on the induction of the EMT and consequent airway remodeling, emphasizing the need for targeted therapeutic strategies to mitigate these effects in patients with CRS.
Hypoxia
In CRS, the blockage of mucus drainage in the sinuses can lead to mucus retention and subsequent hypoxia. Hypoxia in mucus is marked by an increase in HIF-1α and osteopontin, both of which are elevated in CRSwNP. Under hypoxic conditions, human NECs (hNECs) show increased expression of mesenchymal markers (α-SMA, vimentin, Twist) and decreased expression of epithelial markers (E-cadherin, β-catenin). Hypoxia downregulates the catalytic subunit of protein phosphatase-2A (PP2Ac) and upregulates pSmad3, which induces α-SMA. While hypoxia upregulates pSmad3, it is less effective than TGF-β1 alone in inducing the EMT, indicating that HIF-1α alone is insufficient to fully drive the EMT [31]. Additionally, hypoxia activates ten-eleven translocation methylcytosine dioxygenase (TET) 1, a 5-methylcytosine-specific dioxygenase that induces DNA demethylation, acting as a coactivator for HIF-1α and contributing to the EMT [32]. Sirtuin 1 (SIRT1) fine-tunes the cellular response to hypoxia by deacetylating HIF-1α and HIF-2α, leading to the dissociation of p300 from the C-terminal transactivation domain of HIF-1α, thereby inhibiting HIF-1α transcriptional activity [33]. This inhibition of HIF-1α and its downstream target phosphoinositidedependent kinase (PDK)-1 suppresses the HIF-1-induced EMT and prevents the formation of nasal polyps [33,34]. In CRSsNP, the inflammatory environment stimulates SIRT1, which is known to inhibit the HIF-1α-induced EMT. However, in nasal polyp mucosa, there appears to be a loss of SIRT1 activation. Downregulation of miR-155-5p targets SIRT1 to inhibit EMT in primary hNECs [35]. Neural precursor cell expressed developmentally downregulated gene 4-like (NEDD4L) inhibits the EMT by directly degrading β-catenin and HIF-1α or indirectly mediating discoidin domain receptor tyrosine kinase degradation, thereby disrupting the positive feedback loop between β-catenin and HIF-1α [36]. These insights into the role of hypoxia in CRS highlight potential therapeutic targets to mitigate disease progression and improve patient outcomes.
Particulate matter
Particulate matter (PM) disrupts the epithelial barrier through oxidative stress, leading to the degradation of tight junction molecules such as ZO-1, occludin, and claudin-1. Pretreatment with N-acetyl-L-cysteine was found to mitigate PM2.5-mediated reactive oxygen species generation in RPMI 2650 cells, further preventing barrier dysfunction and weakening the degradation of tight junction proteins [37]. In CRS patients, there is an increase in proinflammatory cytokines, tissue inhibitors of metalloprotease-1, and thymic stromal lymphopoietin (TSLP), indicating an inflammatory response to PM exposure [38].
Diesel exhaust particles (DEPs) are an important component of PM. NECs from patients with CRSsNP are more susceptible to the EMT due to immunological memory. Exposure of hNECs to DEP results in larger and more spindle/fibroblast-like shapes. DEP exposure decreases E-cadherin expression and increases protein levels of mesenchymal markers (N-cadherin and α-SMA), while reducing levels of epithelial markers (E-cadherin and ZO-1). The knockdown of ZEB2, an essential transcription factor in EMT, has been found to reverse the DEP-induced EMT, as demonstrated by an increase in epithelial markers (E-cadherin and ZO-1) and a reduction in mesenchymal markers (N-cadherin and α-SMA) [14].
Tobacco smoke
Tobacco smoke induces type 2 inflammatory changes, as evidenced by increased expression of type 2 helper T cell (Th2) cytokines and total serum immunoglobulin E. Current smokers were found to exhibit higher expression of mesenchymal markers (N-cadherin, α-SMA, and vimentin) and lower expression of epithelial markers (E-cadherin) than nonsmokers [39]. Tobacco smoke contributes to the exacerbation of CRS by releasing local epithelial-derived cytokines and Th2 cytokines, thereby inducing the EMT in CRSwNP patients [39]. Cigarette smoke also increases the release of TGF-β1, activates TGF signaling (promoting the phosphorylation of SMAD2 and SMAD3), increases cell migration, and decreases E-cadherin expression [40].
Detergents
Anionic surfactants and detergents have been shown to disrupt tight junctions and associated molecules, compromising the barrier integrity of keratinocytes and bronchial epithelial cells. Significant increases in IL-33 and TSLP in bronchial epithelium suggest that laundry detergents may induce the Th2 immune response [41].
Research on human bronchial epithelial cells cultured at the air-liquid interface has revealed that laundry detergent treatment markedly impairs epithelial barrier function. This impairment is evidenced by a reduction in transepithelial electrical resistance, an increase in paracellular permeability, and deformations in the tight junction structure [42].
Additionally, a study of mice exposed to an industrial bleach and detergent mixture through inhalation showed significant changes in tracheal cell morphology. Notable observations included a reduction in the length of ciliated columnar cells, a decrease in goblet cells, and the loss of cilia. These morphological changes further highlight the adverse effects of detergent exposure on respiratory epithelial cells [43].
EPITHELIAL BARRIER AND THE EPITHELIAL-MESENCHYMAL TRANSITION
The nasal respiratory epithelium is composed of pseudostratified columnar epithelium with ciliated cells, goblet cells, basal cells (BCs), submucosal gland cells, and secretory cells [44]. BCs possess self-renewing capabilities and are responsible for regenerating various differentiated cell types of nasal epithelium to maintain epithelial homeostasis and barrier integrity [45]. In response to epithelial damage, BCs proliferate and migrate to the site of injury, differentiating into ciliated or goblet cells to reconstruct the epithelium and restore barrier function [46]. However, in pathologic conditions, abnormal repair processes may change these differentiation patterns, resulting in failure to properly restore the epithelial barrier [47].
In CRS, epithelial barrier damage initiates repair processes, reactivating the EMT, which is one of the hallmarks of the disease [4]. Early studies on CRS identified defective epithelial barriers in the nasal sinus. Disarray in the nasal epithelial structure in CRS can manifest as acanthosis and acantholysis. Altered states of differentiation and acanthosis in CRS have been proposed to result from a cycle of ongoing injury and repair associated with the EMT. The loss of tight junction proteins such as ZO-1 and E-cadherin can lead to acantholysis [4].
Barrier dysfunction and inflammation are closely linked in CRS. The loss of barrier integrity allows the influx of antigens, irritants, and pathogens, leading to inflammation, which in turn perpetuates barrier loss [1]. Recent studies have elucidated how type 2 inflammatory environments induce the EMT. In patients with CRSwNP, increased levels of Th2 cytokines have been frequently discussed in conjunction with the EMT. IL-4 and IL-13 stimulate the EMT in epithelial cells and are key factors in the regulation of polyp formation [48-50]. Coculture of human nasal epithelial cells with M2 macrophages has been shown to induce the EMT via the IL4/STAT6/interferon regulatory factor 4 signaling pathway [51]. BMP2, which is primarily expressed by eosinophils and macrophages, has been identified as a significant biomarker for refractory CRSwNP and can induce the EMT in a type 2 inflammatory environment [9]. T-cell membrane protein-4, which is upregulated in macrophages from CRSwNP patients, contributes to polyp formation through the TGF-β1-mediated EMT in nasal epithelial cells [52].
Eosinophils contribute to epithelial dysfunction and promote the EMT [48]. Anti-IL5 treatment led to a marked reduction in several EMT markers in the airway epithelium, suggesting that eosinophils and their products are important in barrier dysfunction and EMT induction [53].
The EMT plays a crucial role in nasal tissue remodeling and fibrosis in the context of chronic inflammation [54,55]. During the repair process of epithelial damage, mesenchymal cells can produce collagens and extracellular matrix proteins, causing thickening of the basement membrane and tissue fibrosis, which are well-known phenomena in CRSsNP and non-type 2 CRS [55,56]. The S100A4-induced EMT transforms epithelial cells into fibroblasts that secrete pro-alpha1 chains of type 1 collagen, contributing to extracellular collagen accumulation and tissue remodeling in the nasal sinus [10].
The EMT is frequently observed in nasal polyps of CRSwNP patients. During the early stages of polyp formation, fibroblasts are activated, with more EMT occurring in the stalk than in the body of the polyps [4,54]. In CRSwNP patients, the proportion of epithelial cells is reduced, while the number of fibroblasts increases in nasal polyp tissues, suggesting that the EMT may induce a change in populations from epithelial cells to fibroblasts during polyp formation [54,57,58]. Moreover, recent research has indicated that epithelial cells may have a tendency to transform into fibroblasts during the EMT [10,58].
Single-cell RNA sequencing has revealed that nasal polyps in CRSwNP patients exhibit decreased levels of adhesion G protein-coupled receptor (ADGR) B3-positive fibroblasts and increased periostin (POSTN)-positive fibroblasts [58]. ADGRB3 reduction is associated with decreased cell adhesion, while increased levels of POSTN are associated with EMT promotion in various cancers. Additionally, gene screening indicates that WAP four-disulfide core domain 2, a protease inhibitor critical for colon mucosal homeostasis, is downregulated in nasal polyps, while the type 2 inflammation marker C-C motif chemokine ligand 26 is upregulated. Both genes may contribute to the EMT in CRSwNP [58].
INTRACELLULAR SIGNALING PATHWAYS
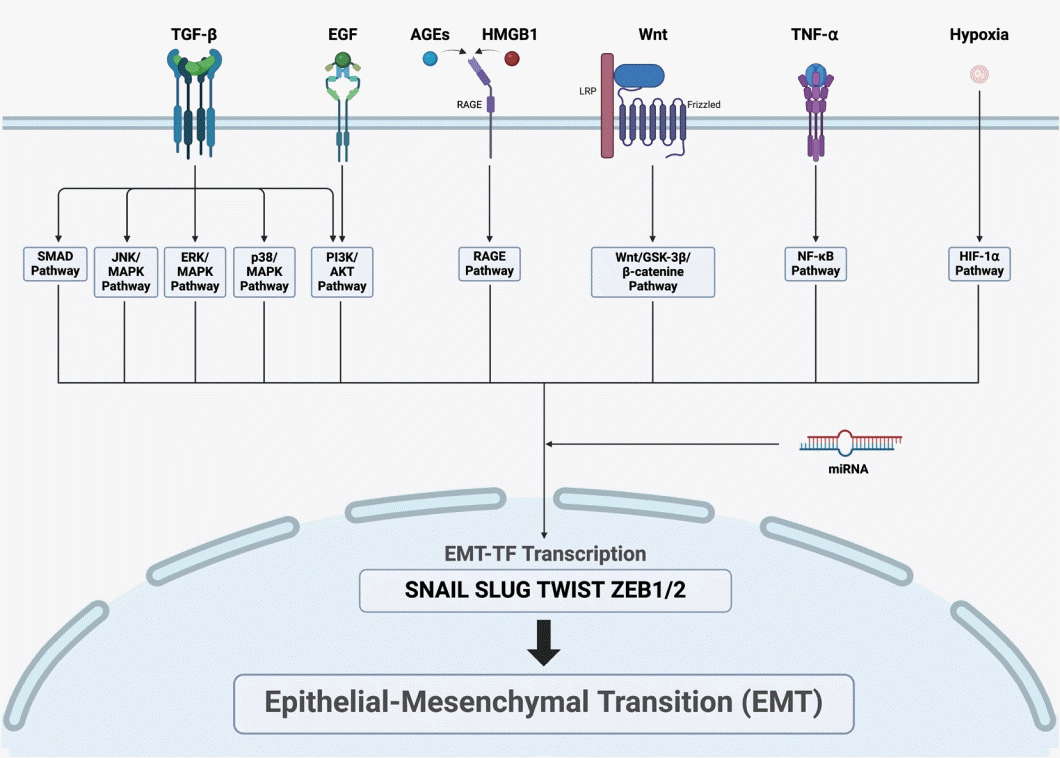
The EMT is initiated with the activation of intracellular signaling pathways, which converge to activate EMT-TFs. Various intracellular signaling pathways that induce EMT have been identified in CRS. These pathways can be triggered by TGF-β, EGF, advanced glycation end-products (AGEs), high mobility group box (HMGB) 1, Wnt, and tumor necrosis factor (TNF)-α (Fig. 2).
TGF-β
TGF-β induces the EMT through the Smad2/3-dependent pathway. Levels of TGF-β1 and Smad3 are significantly higher in CRSsNP patients than in controls. TGF-β receptors, TβRI and TβRII, are also highly expressed in the nasal mucosa of CRSsNP patients [59]. While the upstream signaling pathway of TGF-β is upregulated in CRSsNP patients, overexpression of Smad7 can inhibit downstream components such as pSmad3, exerting an antiproliferative effect on sinonasal epithelium from TGF-β in nasal epithelial cells [60]. In CRSwNP, overexpression of CD109 inhibits the TGF-β-induced EMT through SMAD2/3-p [61]. Inhibition of TBX1 blocks the TGF-β-Smad 2/3 pathway, thereby suppressing the EMT [62]. TGF-β was found to decrease levels of miR-203a-3p and increase levels of sine oculis homeobox homolog (SIX) 1 in a treatment time-dependent manner, with miR-203a-3p regulating the Smad3 pathway by inhibiting SIX1 to suppress the EMT [63].
TGF-β1 induces DNMT expression in a dose-dependent manner, leading to DNA methylation and EMT through the p38, c-Jun N-terminal kinase, Snail, and Slug signaling pathways [64]. It also activates phosphoinositide 3-kinase, which in turn activates protein kinase B (AKT), promoting the EMT [65]. Additionally, TGF-β activates the extracellular signalregulated kinase (ERK), p38, and JUN N-terminal kinase mitogen-activated protein kinase (MAPK) pathways [66]. Heat shock protein (HSP) 47, which is involved in collagen maturation and ECM remodeling, is upregulated by TGF-β1 in a dose- and time-dependent manner. An miR-29b mimic was found to significantly inhibit the expression of HSP47 and TGF-β1-induced EMT markers by directly binding to the HSP47 target site [67].
In CRSwNP, the expression of E-cadherin is downregulated, whereas TGF-β1, α-SMA, fibronectin, and vimentin are upregulated compared to CRSsNP and control individuals. miR-21 and TGF-β1 mRNA levels are significantly higher in CRSwNP, with the TGF-β1/miR-21/phosphatase and tensin homolog (PTEN)/Akt axis contributing to pathogenesis by upregulating miR-21, reducing PTEN, and increasing Akt phosphorylation [68]. TGF-β1 also activates histone deacetylase (HDAC) 2/4 to induce the EMT, with trichostatin A inhibiting HDAC 2/4 to reduce the EMT [69].
TGF-β induces stress in the endoplasmic reticulum, leading to the unfolded protein response, which targets c-Src to induce the EMT. Increased Src activity promotes the EMT, while c-Src inhibition suppresses it [70].
HMGB1
HMGB1, a ligand for the receptor for advanced glycation end-products (RAGE), is upregulated in refractory CRSwNP and correlates with disease severity [71]. In a dose-dependent manner, HMGB1 upregulates N-cadherin and vimentin while downregulating ZO-1 and E-cadherin in epithelial cells isolated from ECRSwNP. Hypoxia-induced HMGB1 promotes EMT through the RAGE signaling pathway [72]. TET 2 activates peroxisome proliferator-activated receptor-γ, which subsequently inhibits HMGB1, thereby suppressing the EMT [73,74]. Upregulation of miR-1287-5p inhibits IL-6, IL-8, TNF-α, and the EMT by targeting and suppressing Snail family transcriptional repressor (SNAI) 1 and HMGB1. Knockdown of SNAI1 reduces HMGB1, reducing the levels of proinflammatory cytokines. Conversely, HMGB1 inhibitors decrease levels of SNAI1, thereby suppressing the EMT process [75]. These findings underscore the critical role of HMGB1 in the pathogenesis of CRSwNP, particularly through its regulation of the EMT and the associated inflammatory responses.
Wnt
Wnt-related molecules such as β-catenin, WNT3A, and cyclin D1 show upregulation in CRSwNP patients. In a study using a CRS mouse model, activation of Wnt signaling induced more polyp lesions, implying that activation of the Wnt pathway contributes to the inflammatory environment and tissue remodeling in CRSwNP [76]. The Wnt/glycogen synthase kinase-3β/β-catenin/transcription factor-4 pathway leads to the transcription of S100A4, inducing the EMT [10]. The Wnt pathway promotes β-catenin translocation to the nucleus, inhibiting its binding to E-cadherin at the epithelial cell membrane, thereby weakening epithelial cell adhesion and affecting the EMT process [10].
AGE/ERK
Studies have shown that the AGE/RAGE/ERK pathway promotes EMT and tissue remodeling in CRSwNP. The AGE/RAGE complex maintains the transition of epithelial cells to mesenchymal cells and activates the ERK pathway, promoting stromal tissue edema and tissue remodeling [77]. The interaction between AGEs and RAGE proceeds through p38 MAPK and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling, as demonstrated in pelvic organ prolapse [78]. miR-200a-3p inhibits p38/ERK, suppressing ZEB1 and consequently inhibiting the EMT [13].
In patients with neutrophilic CRS, the ERK pathway is activated by high interferon (IFN)-γ expression, correlating with the induction of EMT markers [79]. IFN-γ promotes the EMT in human nasal epithelial cells through the JAK/STAT1/interferon consensus sequence-binding protein/p38 and ERK signaling pathways. Expression levels of p-ERK and p-p38 increase with CRS progression independently of the HIF-1α, SMAD, and NF-κB signaling pathways [79].
TNF-α
TNF-α augments the effects of TGF-β1 on the EMT through a Smad-dependent mechanism, independent of extracellular ERK activation. TNF-α alone can reduce E-cadherin expression by approximately 50% [80]. TNF-α and IL-1β induce the EMT in normal and cancerous breast epithelial cells via NF-κB-mediated upregulation of Twist [81,82].
miRNA
miRNAs play critical roles in regulating the EMT in CRS. miR-30a-5p inhibits the EMT by targeting cyclin-dependent kinase 6 [83]. Upregulation of miR-1287-5p suppresses IL-6, IL-8, TNF-α, and the EMT by targeting SNAI1 and HMGB1, with SNAI1 knockdown reducing levels of HMGB1 and proinflammatory cytokines [75]. miR-451a was found to inhibit the EMT by targeting cadherin 11 in an asthma mouse model [84]. Overexpression of miR-335-5p has anti-inflammatory effects in CRS, interacting with the targeting protein for Xklp 2 to inhibit the Akt signaling pathway and reduce inflammatory cytokine release [85]. Knockdown of miR-761 exacerbates CRS symptoms by stimulating inflammatory responses, while its overexpression deactivates the lipocalin-2/Twist1 signaling pathway, modulating EMT markers [86]. Altered expression levels of miR-34 and miR-449 significantly impair epithelial cilia function in CRSwNP patients [87]. Upregulated miR-125b in airway epithelial cells can promote type I IFN expression by inhibiting 4E-BP1, potentially contributing to mucosal eosinophilia in cases of eosinophilic CRSwNP [88]. TGF-β1 induces the EMT through miR-182 [68].
CONCLUSION
CRS is a complex condition characterized by prolonged inflammation of the nasal and sinus mucosa. It is divided into various phenotypes, initially distinguished by the presence or absence of nasal polyps, and further categorized by their inflammatory signatures. This disease demonstrates significant variability in clinical presentation and underlying immunopathology, which are influenced by geographic variations, microbial colonization, and environmental exposures.
The EMT plays a crucial role in the progression of CRS by compromising the integrity of the epithelial barrier, which leads to tissue remodeling and fibrosis. In CRS, this barrier is often impaired, heightening vulnerability to external factors like pathogens, allergens, and environmental pollutants. Such disruptions trigger repair mechanisms that involve EMT, thereby sustaining a cycle of injury and impaired healing.
Key intracellular signaling pathways regulate the EMT in CRS, including TGF-β, HMGB1, Wnt, AGE/ERK, TNF-α, and various miRNAs. These pathways play a pivotal role in controlling cell adhesion, migration, and extracellular matrix production, which are instrumental in driving the pathological changes observed in CRS. The EMT involves transcriptional reprogramming that is regulated by key transcription factors such as Snail, Slug, Twist, and ZEB1/2, as well as signaling pathways including TGF-β, Wnt, and Notch. This process leads to the loss of epithelial markers and the acquisition of mesenchymal traits. Additionally, the disruption of tight junction proteins and cytoskeletal alterations further compromise barrier function and facilitate chronic inflammation.
The association between EMT and CRS highlights potential therapeutic targets that focus on maintaining epithelial integrity and reducing inflammation. By modulating key signaling pathways and transcription factors, novel strategies for preventing or treating EMT can be developed, which may address the chronicity and severity of CRS. Additionally, elucidating the role of environmental and microbial factors in promoting EMT could lead to preventive measures and therapeutic approaches that are customized to the needs of individual patients.
Future research should aim to elucidate the precise molecular mechanisms underlying the EMT in CRS, exploring the interplay between genetic predisposition, environmental triggers, and immunological responses. Targeting the key regulators and processes involved in the EMT, may make it possible to develop treatments that not only alleviate symptoms but also address the root causes of chronic inflammation and tissue remodeling.
In conclusion, the intricate relationship between epithelial barrier integrity, the EMT, and chronic inflammation is important for the pathogenesis of CRS. Advances in understanding these processes offer promising avenues for therapeutic intervention, highlighting the potential for targeted treatments that restore epithelial function and prevent chronic inflammation and tissue remodeling in CRS.
 XML Download
XML Download