

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
There has recently been growing interest in the adverse impacts of air pollution on human health. Particulate matter (PM), one of key components of air pollution, is categorized into PM10 (diameter smaller than 10 μm), PM2.5 (diameter smaller than 2.5 μm), and PM0.1 (diameter smaller than 0.1 μm) according to its aerodynamic diameter [1,2]. The health effects of PM vary depending on its size; smaller particles can penetrate and accumulate in the lower respiratory tract, and particles smaller than 1 μm can even diffuse into the circulation through the alveoli and deposit in cell tissue [3-5]. Numerous studies have explored the detrimental health impacts of PM, which include cardiovascular, respiratory, neurological, and reproductive system diseases [6-11]. PM generates oxidative stress through the production of reactive oxygen species (ROS), leading to inflammatory responses and cell death [12-15]. It can trigger cell death in various organs via multiple pathways, including necrosis, apoptosis, autophagy, pyroptosis, and ferroptosis [16]. Among the various patterns of cell death, pyroptosis is a relatively recently discovered inflammatory cell death process characterized by nuclear condensation, cell swelling, and plasma membrane rupture without DNA fragmentation [17]. Various stimuli, including pathogenassociated molecular patterns and damage-associated molecular patterns, induce the formation of the NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasome complex. This complex generates mature caspase-1, which produces the pro-inflammatory cytokines interleukin (IL)-1β and IL-18 by cleaving pro-IL-1β and pro-IL-18, respectively. Additionally, it triggers the cleavage of gasdermin D (GSDMD), leading to the formation of GSDMD-N. GSDMD-N binds to phospholipids on the plasma membrane, forming pores that induce pyroptosis [17,18]. Some studies have suggested that PM induces tissue injury through pyroptosis, demonstrating its role in conditions such as neuronal injury, myocardial injury, renal tubular injury, lung injury, and corneal injury [9,19-22]. However, few attempts have been made to investigate pyroptosis induced by PM in nasal epithelial cells. This study investigated PM-induced pyroptosis and the underlying signaling mechanisms in human nasal epithelial cells.
METHODS
Ethics
The study protocol was approved by the Institutional Review Board of Daejeon St. Mary’s Hospital (IRB policy No. DC15TISI0022, 22 May 2020).
Reagents
Antibiotic-antimycotic solution, fetal bovine serum (FBS) solution, and Eagle’s Minimum Essential Medium (EMEM) were sourced from Gibco, a brand of Thermo Fisher Scientific (Waltham, MA, USA). The lactate dehydrogenase (LDH) assay, IL-1β and IL-18 enzyme-linked immunosorbent assay (ELISA) kits, and 4',6-diamidino-2-phenylindole (DAPI) were purchased from Abcam (Cambridge, UK). The PM standard reference material SRM 2786, MCC950, and propidium iodide (PI) were obtained from Millipore Sigma-Aldrich (Saint Louis, MO, USA). Antibodies specific for NLRP3, glyceraldehyde phosphate dehydrogenase (GAPDH), and cleaved caspase-1 were procured from Cell Signaling Technology (Danvers, MA, USA). Additionally, the antibody targeting GSDMD-N was acquired from Abcam.
Preparation of the PM solutions
The PM standard reference material SRM 2786 (Millipore Sigma-Aldrich, St. Louis, MO, USA) was dispersed in phosphate-buffered saline (PBS) to achieve a final concentration of 100 μg/mL. It was then further diluted to 50 μg/mL and 25 μg/mL and subjected to 30 minutes of ultrasonication prior to treatment. These concentrations were selected based on the study conducted by Wang et al. [23].
Cell culture
Human nasal epithelial RPMI-2650 cells (ATCC#CCL-30, ATCC, Manassas, VA, USA) were cultured in EMEM (ATCC# 30-2003) supplemented with 1% antibiotic-antimycotic solution and 10% FBS. The culture medium was refreshed every 2–3 days to maintain cell integrity and provide essential nutrients. The cells were incubated in a humidified environment to ensure optimal conditions for growth.
Detection of cell viability
To assess cell viability and cytotoxicity, we performed a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay using an EZ-Cytox Cell Viability Assay Kit (DoGen, Seoul, Republic of Korea). We applied 10 μL of the MTT assay kit reagent to each well containing the cells after the designated treatment. The plate was then incubated for an additional 2 hours to allow the conversion of MTT into formazan crystals by viable cells. Subsequently, the absorbance of the formazan product was measured at a wavelength of 450 nm using a microplate reader (Bio-Rad, Hercules, CA, USA).
Cell death analysis (PI and LDH release)
To evaluate pore formation in cell membranes, PI staining was conducted. Following the treatment, the cells were stained with a PI mixture and incubated for 25 minutes at 37°C. The stained cells were subsequently observed and imaged using a fluorescence microscope. To assess cell membrane damage, LDH release was measured. Cell culture supernatants were collected after the specified treatment, and LDH release was quantified using an LDH Kit according to the manufacturer’s guidelines.
Western blot
To extract proteins, cells were rinsed with PBS and lysed using radioimmunoprecipitation assay buffer (Elpis Biotech, Daejeon, Korea) supplemented with protease inhibitors (Roche Diagnostics, Mannheim, Germany). The lysates were then centrifuged for 15 minutes at 13,000 rpm to isolate the protein fraction. Protein concentration was determined using a bicinchoninic acid protein assay kit from Pierce (Rockford, IL, USA). Equal amounts of proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes (Bio-Rad). The membranes were incubated overnight at 4°C with primary antibodies against NLRP3 (1:500), GSDMD-N (1:500), caspase-1 (P20, 1:500), and GAPDH (1:1,000). Subsequently, the membranes were treated with horseradish peroxidase-conjugated secondary antibody and the signals were developed using an enhanced chemiluminescence reaction. Positive bands were detected using a ChemiDoc XRS+ system (Bio-Rad).
Immunofluorescence staining
After rinsing the cells with PBS, they were fixed with a 4% paraformaldehyde solution for 15 minutes. The cells were then permeabilized with a 0.2% Triton-X-100 solution at room temperature for 15 minutes. Subsequently, the cells were blocked with 5% bovine serum albumin for 1 hour to prevent nonspecific antibody binding and reduce background signal. The primary antibody against caspase-1 (P20) was added at a 1:200 dilution, and the cells were incubated overnight at 4°C. Following another wash, the cells were treated with an antirabbit Alexa Fluor 488 secondary antibody at a 1:1,000 dilution and incubated in the dark for 1 hour at room temperature. To visualize the nuclei, the cells were stained with DAPI for 10 minutes. The stained cells were examined using an Olympus IX73 fluorescence microscope (Olympus Corp., Tokyo, Japan). For quantitative analysis of immunofluorescence intensity, the images were processed using ImageJ software (version 1.54g; National Institutes of Health, Bethesda, MD, USA).
Enzyme-linked immunosorbent assay
IL-1β and IL-18 protein levels in the culture supernatant were measured using ELISA kits, following the manufacturer’s instructions. The concentrations of IL-1β and IL-18 were determined by correlating the absorbance values with a standard curve, which was created using known concentrations of recombinant cytokines.
Pretreatment with MCC950
To demonstrate that PM-induced pyroptosis occurs via the NLRP3 inflammasome-dependent pathway, cells were first pretreated with 1 μM MCC950, an inhibitor of the NLRP3 inflammasome, for 2 hours. Subsequently, they were exposed to the lowest concentration of PM that demonstrated a significant difference from the control group in a western blot analysis. This was followed by an additional 24-hour incubation period. The 1 μM concentration of MCC950 used in this study was based on the dosage reported in the paper by Roseborough et al. [24].
Statistical analysis
All experiments were conducted independently three times, with one representative experiment displayed. Data analysis was carried out using GraphPad Prism 5 software (GraphPad, Inc., La Jolla, CA, USA). Depending on the experimental design, the statistical significance of differences between control and experimental values was assessed using either the unpaired t-test or one-way analysis of variance (ANOVA). All data were presented as mean±standard error of the mean (SEM).
RESULTS
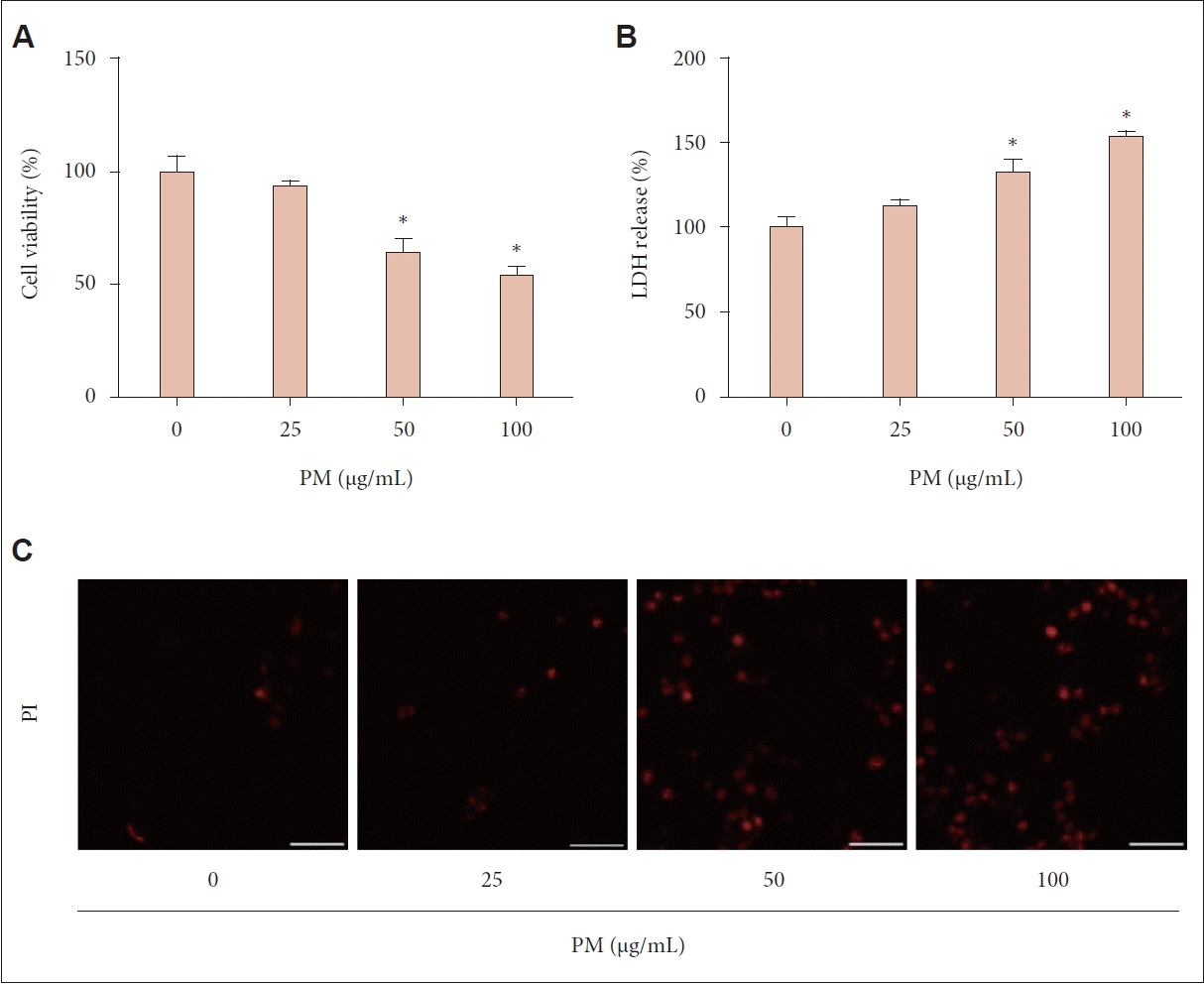
Effect of PM on the viability of human nasal epithelial cells
Cell viability was assessed using the MTT assay after a 24-hour exposure to various PM concentrations (0, 25, 50, or 100 μg/mL). We observed significant reductions in cell viability at PM concentrations of 50 μg/mL or higher, compared to the control group (p<0.05) (Fig. 1A). Similarly, LDH release, which indicates cell death, increased significantly at PM concentrations of 50 μg/mL or higher. This increase in LDH release was more pronounced at higher PM concentrations (p<0.05) (Fig. 1B), suggesting a dose-dependent increase in cell death. Additionally, PI staining, used to confirm cell death, showed higher staining intensity at higher PM concentrations, as observed through fluorescence microscopy (Fig. 1C).
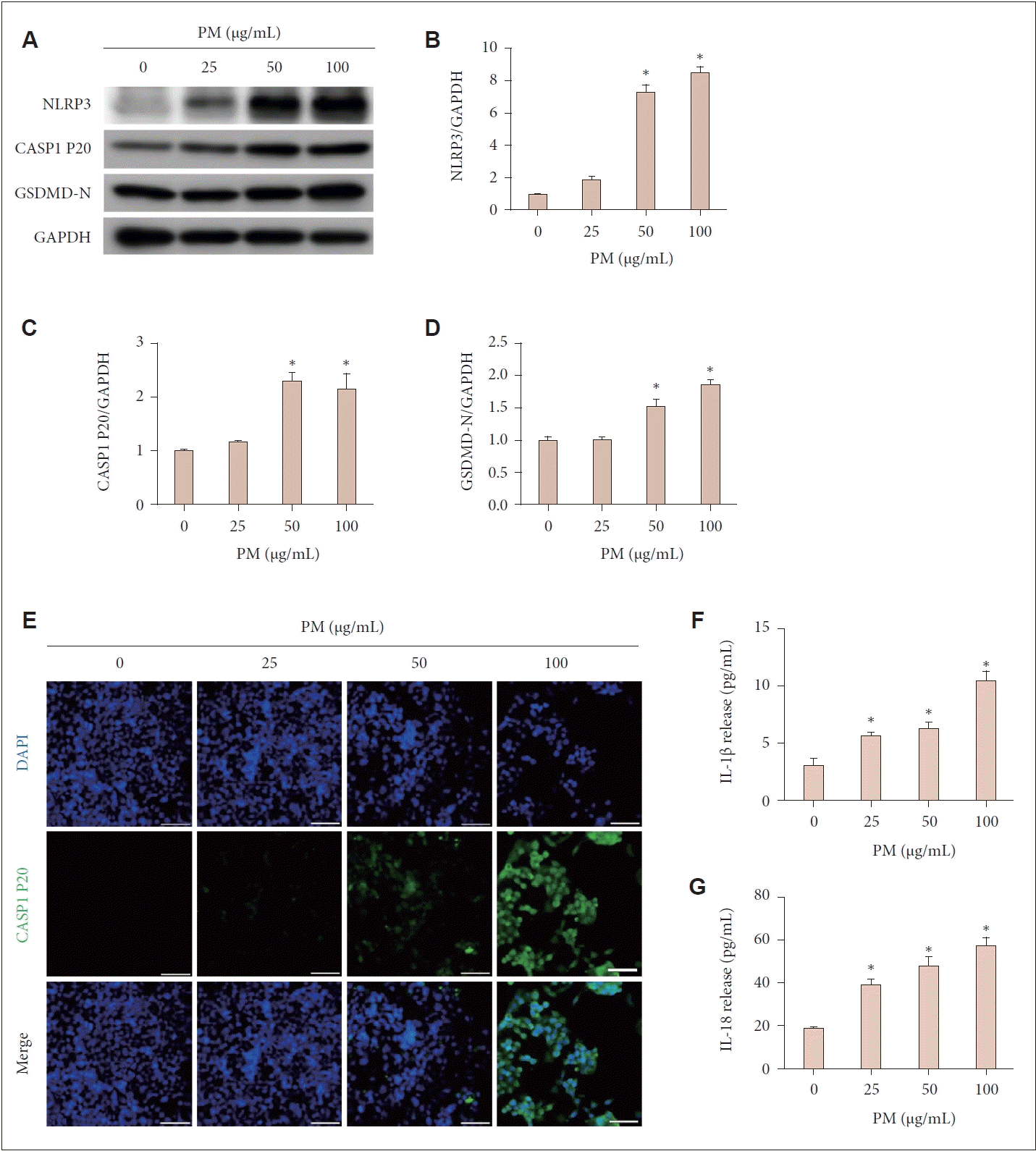
Pyroptosis in human nasal epithelial cells induced by PM
After exposing the cells to various PM concentrations (0, 25, 50, or 100 μg/mL) for 24 hours, we conducted western blot analysis to evaluate the expression levels of key proteins involved in NLRP3 inflammasome activation: cleaved caspase-1 (CASP1 P20), NLRP3, and GSDMD-N (Fig. 2A). The protein levels of NLRP3, CASP1 P20, and GSDMD-N, relative to the GAPDH control, were significantly elevated at PM concentrations of 50 μg/mL or higher compared to the control (p<0.05) (Fig. 2B-D). Additionally, immunofluorescence staining showed increased expression levels of CASP1 P20 with rising PM concentrations (Fig. 2E). ELISA analysis of the cell culture supernatants indicated a significant increase in IL-1β and IL-18 protein levels at PM concentrations of 25 μg/mL or higher compared to the control, with the intensity of release increasing as PM concentration rose (p<0.05) (Fig. 2F and G). In summary, these findings suggest that PM induces pyroptosis in human nasal epithelial cells.
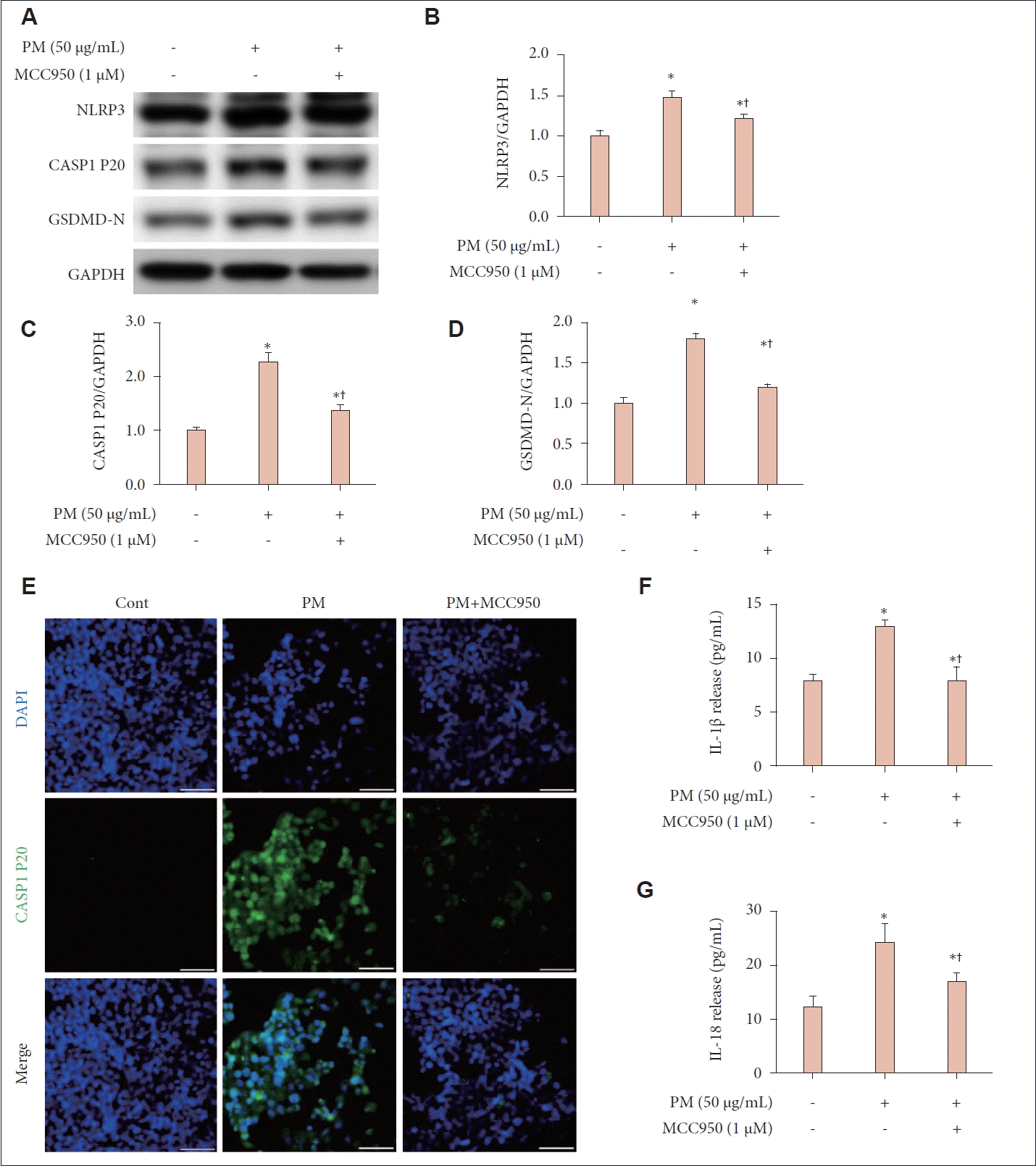
PM provoked pyroptosis of human nasal epithelial cells in an NLRP3 inflammasome-dependent manner
Cells were pretreated with 1 μM MCC950, an NLRP3 inflammasome inhibitor, for 2 hours before being exposed to 50 μg/mL of PM, followed by an additional 24-hour incubation. The increased western blot signals of CASP1 P20, NLRP3, and GSDMD-N observed in cells exposed to 50 μg/mL of PM, compared to the control, were reversed following the addition of MCC950 (Fig. 3A). Pretreatment with MCC950 led to a significant reduction in the expression levels of NLRP3, CASP1 P20, and GSDMD-N compared to treatment with PM alone (p<0.05) (Fig. 3B-D). Immunofluorescence staining further demonstrated decreased expression levels of CASP1 P20 in samples treated with both MCC950 and PM compared to those treated with PM alone (Fig. 3E). ELISA analysis indicated that the elevated release of IL-1β and IL-18 observed in samples treated with 50 μg/mL of PM was significantly reduced following the addition of MCC950 (p<0.05) (Fig. 3F and G).
DISCUSSION
In this study, we found that PM induces cell death in cultured human nasal epithelial cells through the process of pyroptosis, characterized by a significant increase in the protein levels of NLRP3, CASP1 P20, GSDMD-N, IL-1β, and IL-18. Pretreatment with MCC950, an inhibitor of the NLRP3 inflammasome, resulted in decreased levels of these proteins, suggesting that PM-triggered pyroptosis is mediated by the NLRP3 inflammasome-dependent pathway.
Ren et al. [25] found that PM2.5 initiated NLRP3 inflammasome-mediated pyroptosis by generating ROS in both murine macrophage cells in vitro and mouse lung tissue in vivo. They demonstrated that the addition of the ROS scavenger N-acetyl cysteine led to a reduction in proteins associated with pyroptosis, indicating that ROS generation triggers pyroptosis induced by PM2.5. Similarly, Li et al. [22] reported that exposure to PM2.5 activated the NLRP3/caspase-1 signaling pathway, causing macrophage pyroptosis and subsequent inflammation and injury in the lung tissue of a mouse model. Zheng et al. [26] also confirmed the activation of the NLRP3 inflammasome pathway by PM2.5 through mechanisms such as ROS production, K+ efflux from plasma membrane perturbation, and cathepsin B release due to lysosomal damage, using human monomyelocytic leukemia cell lines. Additionally, they showed that PM2.5 induces pulmonary fibrosis in mice. In research focusing on the upper respiratory tract, Li et al. [27] demonstrated that PM2.5 exacerbated allergic rhinitis symptoms and triggered pyroptosis through NLRP3, caspase-1, and GSDMD-dependent pathways, using an allergic rhinitis mouse model. Another study by Li et al. [28] showed that IL-17A mediated pyroptosis in human nasal epithelial cells via the extracellular signal-regulated kinase (ERK)-NLRP3/caspase-1 pathway, although it did not explore the effects of PM.
Our study, in line with the above findings, appears to be the first to specifically demonstrate that PM induces NLRP3 inflammasome-driven pyroptosis in cultured human nasal epithelial cells. To our knowledge, this has not been previously reported in the literature, marking our research as a novel contribution to understanding the effects of PM on this particular cell type. The use of MCC950, a specific NLRP3 inflammasome inhibitor, provides further insight into our findings. MCC950’s ability to attenuate PM-induced pyroptosis underscores the critical role of the NLRP3 inflammasome pathway in this process. This finding not only supports existing studies on the role of the NLRP3 inflammasome in PM-triggered pyroptosis, but also suggests potential therapeutic interventions targeting this pathway to mitigate the detrimental health effects of PM exposure.
However, there are some limitations to our study. SRM 2786, the PM standard reference material used, contains a variety of organic and inorganic components of different sizes, including PM2.5 and PM10 [29]. Therefore, it is unclear which specific component is the causative agent of NLRP3 inflammasomemediated pyroptosis. Additionally, since cell death induced by PM can occur through various pathways such as necrosis, apoptosis, autophagy, pyroptosis, and ferroptosis, the observed decrease in cell viability may not be solely attributable to pyroptosis, as other pathways may also contribute. Consequently, it was impossible to quantify the specific contribution of pyroptosis to cell death [16]. Lastly, due to the in vitro nature of this study, we were unable to determine how PM-triggered pyroptosis would occur in human nasal epithelial cells under actual in vivo conditions. Future studies are therefore warranted to investigate whether PM causes pyroptosis in human nasal epithelial cells in vivo and to determine which particle composition and size are responsible for pyroptosis. Based on these findings, we anticipate that future research will explore the therapeutic potential of compounds targeting pyroptosis signaling pathways to alleviate the adverse effects of PM exposure on human health.
 XML Download
XML Download