

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Polyomavirus (PV) infection commonly occurs in childhood, with the seroprevalence of BK virus (BKV) reaching up to 90% in adults [1]. BKV typically remains dormant, primarily in kidney tubular epithelial and urothelial cells; however, reactivation of BKV may occur due to immunosuppression in kidney transplant recipients. BK viruria is found in 30%–40% of patients, while BK viremia is identified in 16% to 67% of kidney transplant recipients depending on the definition of viremia based on plasma viral copy number; this typically occurs in the first year after transplantation, with the peak incidence at 3 to 6 months following engraftment. However, up to 18% of cases may occur more than 1 year following transplantation in adults [2–8]. In 10%–20% of individuals with PV reactivation, there is infection with both BKV and JC virus (JCV), while JCV infection alone is rare, occurring in <5% of cases [7,9]. The overall incidence of BKV nephropathy (BKVN) in the first year after transplantation is 10% on average, with the rate of kidney dysfunction or graft loss reaching 51% in this patient cohort [10–14]. Among patients with a viral copy number >10,000 per milliliter of plasma (copies/mL) and no therapeutic intervention, 10% to 50% develop BKVN within 8 to 16 weeks [6,15]. There are no effective treatments for BKVN, with the reduction of immunosuppression being the mainstay of therapy. However, reducing immunosuppression increases the risk of cell-mediated and/or antibody-mediated rejection, with most studies reporting rejection in up to 15% of those with BKVN [15]. Additionally, the histologic features of rejection overlap with those of BKVN, making the diagnosis of rejection challenging in these patients. This article reviews the diagnosis and histopathologic features and classifications of BKVN/polyomavirus nephropathy (PVN), and compares these to the pathology of rejection in kidney transplant recipients.
PATHOGENESIS OF POLYOMAVIRUS NEPHROPATHY
PVs are a family of icosahedral, nonenveloped double-stranded DNA viruses, including BKV, JCV, and simian virus 40 (SV40). BKV is known to have infected approximately 90% of the worldwide population, usually during childhood, and persists in a dormant stable state in kidney tubular and urothelial cells. The virus may reactivate following immunosuppression, with the overall degree of immunosuppression being the most consistent risk factor identified for developing BKVN. Both innate and adaptive cellular immunity work to inhibit virus reactivation, with humoral immunity playing a lesser role. Transplant recipients with fewer peripheral blood dendritic cells, a muted natural killer cell response, and increased innate immune mediators are at a higher risk of developing BKVN [16–18]. The adaptive immune response, in particular involving BKV-specific T cells, is critical for viral clearance [19,20]. The T cell response to BKV is mediated predominantly by CD4+ T cells, which likely have a direct role in controlling BKV infection. Other adaptive immune responses are mediated through the expression of proinflammatory cytokines, including interferon gamma, which may regulate BKV lytic infection [18,21–23]. It has been suggested that the use of tacrolimus-mycophenolate mofetil (vs. cyclosporine-mycophenolate mofetil) and lymphocyte-depleting medications increases the risk of developing viral nephropathy, possibly via a reduction in interferon secretion [24–26]. Humoral immunity is characterized by a BKV-specific antibody response, which may play a role in neutralizing the circulating virus. However, antibodies alone are unable to control persistent latent infection or provide protection against BKVN [18,27].
BKVN most often occurs due to BKV transported in the donor kidney tubular epithelial and/or urothelial cells. However, determining whether the causative virus is of donor or recipient origin requires knowledge of the donor and recipient BKV subtypes prior to transplantation and the subtype of the virus causing BKVN [28–30]. Publications have reported risk factors for BKVN in kidney transplant recipients, although these risk factors vary somewhat across different studies. Donor risk factors include BKV seropositivity at the time of transplantation, a deceased donor, female sex, older age, and possibly Afro-Caribbean ethnicity and the absence of human leukocyte antigen (HLA) C7. Recipient risk factors include older age in adults and younger age in children, male sex, obesity, diabetes, ureteral stent placement, high serum albumin, and a low BKV antibody titer, direct bilirubin level, and neutrophil count. Transplant factors associated with developing BKVN include repeat kidney transplant, prior rejection episodes, HLA mismatch, ABO incompatibility, and prolonged cold ischemia time [28,31–36].
DIAGNOSIS OF POLYOMAVIRUS NEPHROPATHY
Approximately 20 years ago, the diagnosis of BKVN was categorized as either presumptive/probable, based on evidence of significant viral replication—with varying definitions for this in the literature—or as proven, requiring histologic proof of intrarenal PV infection. A review of these definitions is provided by Imlay et al. [37] and is discussed briefly in Table 1.
Clinical Diagnosis of Polyomavirus Nephropathy
The clinical diagnosis of BKVN utilizes measurements of BKV DNA in the urine and/or plasma using quantitative real-time polymerase chain reaction. Approximately half of kidney transplant recipients with high-level viruria develop BKV viremia within 6 weeks, and of these patients, up to 50% are diagnosed with BKVN. In 2013 and again in 2019, Hirsch and Randhawa [32,38] for the American Society of Transplantation (AST) proposed a definition of possible PVN using urine. This included the presence of decoy cells (virally infected tubular epithelial or urothelial cells, usually by BKV, showing nuclear inclusions), BKV DNA at >1,000,000 copies/mL, or PV clusters identified by electron microscopy using negative staining (Haufen) [32,38]. However, there are substantial physiological fluctuations in urinary viral loads, and the positive predictive value of urinary findings for BKVN is quite low [32,45,46]. Therefore, the concept of possible BKVN was not included in a 2022 consensus definition [37]. Nevertheless, urine screening is noninvasive and has a high negative predictive value. Therefore, the detection of three decoy cells per high power field or urine BKV DNA levels greater than 1,000,000 copies/mL continues to be used as a screening criterion to prompt further assessment for viremia.
Probable/Presumptive BK Virus Nephropathy
Multiple studies have identified a plasma BKV level of >10,000 copies/mL as correlating with development of BKVN [31,39,46–48]. Elfadawy et al. [49] found there was a greater risk for BKVN and subsequent graft dysfunction when this level of viremia was persistently present for 3 or 4 weeks. This criterion for the plasma viral load was adopted by the AST for the presumptive diagnosis of BKVN and deemed sufficient to reduce the level of immunosuppression. However, some investigators have suggested that this level of BKV DNAemia is too high and misses a significant number of patients with BKVN [48]. The AST guidelines published in 2019 acknowledged this and defined probable and presumptive BKVN as 1,000 copies/mL plasma found in two measurements within 3 weeks, and >10,000 copies/mL in at least one of two measurements within 3 weeks, respectively [38]. Subsequently, in 2022, the BK Disease Definitions Working Group of the Transplantation Virus Infection Forum published consensus definitions of PV infection and PVN, including clinical and laboratory findings, to promote consistency in patient recruitment for clinical trials and research [37]. In this iteration, the diagnosis of probable/presumptive PVN requires current treatment with immunosuppression, significant viremia on repeated measures (with specific levels not provided), kidney allograft dysfunction with a >20% rise in the serum creatinine level from baseline, no other process to explain the increased serum creatinine, and no or inadequate kidney biopsy.
While it is clinically useful to have specific plasma viral levels for the probable or presumptive diagnosis of BKVN, the detection of BKV DNA has not been standardized. Preanalytic and analytic limitations impact the reliability and consistency of viral copy number measurements across assays. These include preanalytic variability in biologic samples, such as infection with JCV, the fact that shed BKV DNA does not necessarily reflect active viral replication, and the possible detection of BKV from nonkidney sites. Different laboratories may have analytic differences in DNA extraction techniques, primer and probe sequences used, and polymerase chain reaction cycling parameters, and the potential identification of shed inactive nonreplicating viral DNA is also a meaningful limitation [34,38,50]. The World Health Organization has established an international standard for viral load values expressed as international units/mL; however, there remains variability among laboratories due to the above factors [51–53]. To address these concerns, it has been suggested that each laboratory determine significant levels by validating their assay with kidney biopsy findings of BKVN. However, as a body of literature supports >10,000 copies/mL plasma measured twice 3–4 weeks apart as correlating with biopsy-confirmed BKVN, this appears to be a reasonable standard to use in routine clinical practice until more data or standardized testing becomes available.
Several studies have addressed the accuracy of the presumptive/probable diagnosis of BKVN. Shen et al. [6] reported that viremia of >10,000 copies/mL had a positive predictive value of 50%–82%, a negative predictive value of 100%, a sensitivity of 100%, and a specificity of 88%–96%. Other studies have reported a positive predictive value for BKVN of >90% in the settings of very high viral loads of >1,000,000 copies/mL plasma, BKV noncoding control region (NCCR) gene rearrangements in the blood, or kidney functional decline of >20% from baseline [38,54,55]. A study of 213 kidney transplant recipients demonstrated that a plasma viral load of >92,850 copies/mL was able to predict BKVN with 89% sensitivity and 94.6% specificity [5]. Peak viral loads of >185,000 copies/mL plasma on the first positive test or of 223,000 copies/mL at any time have been associated with odds ratios of 113.25 (P<0.001) and 70.5 (P=0.0001), respectively, for the development of BKVN [56]. Therefore, while the predictive value of specific levels of viremia for BKVN has not been established definitively, the value currently recommended for considering immunosuppression reduction is >10,000 copies/mL, particularly when this level has been persistently present [57]. Interestingly, Elfadawy et al. [49] found that even though transiently high viremia was not associated with BKVN, it was associated with overall worse graft function.
Proven/Histopathologic Diagnosis of BK Virus/Polyomavirus Nephropathy
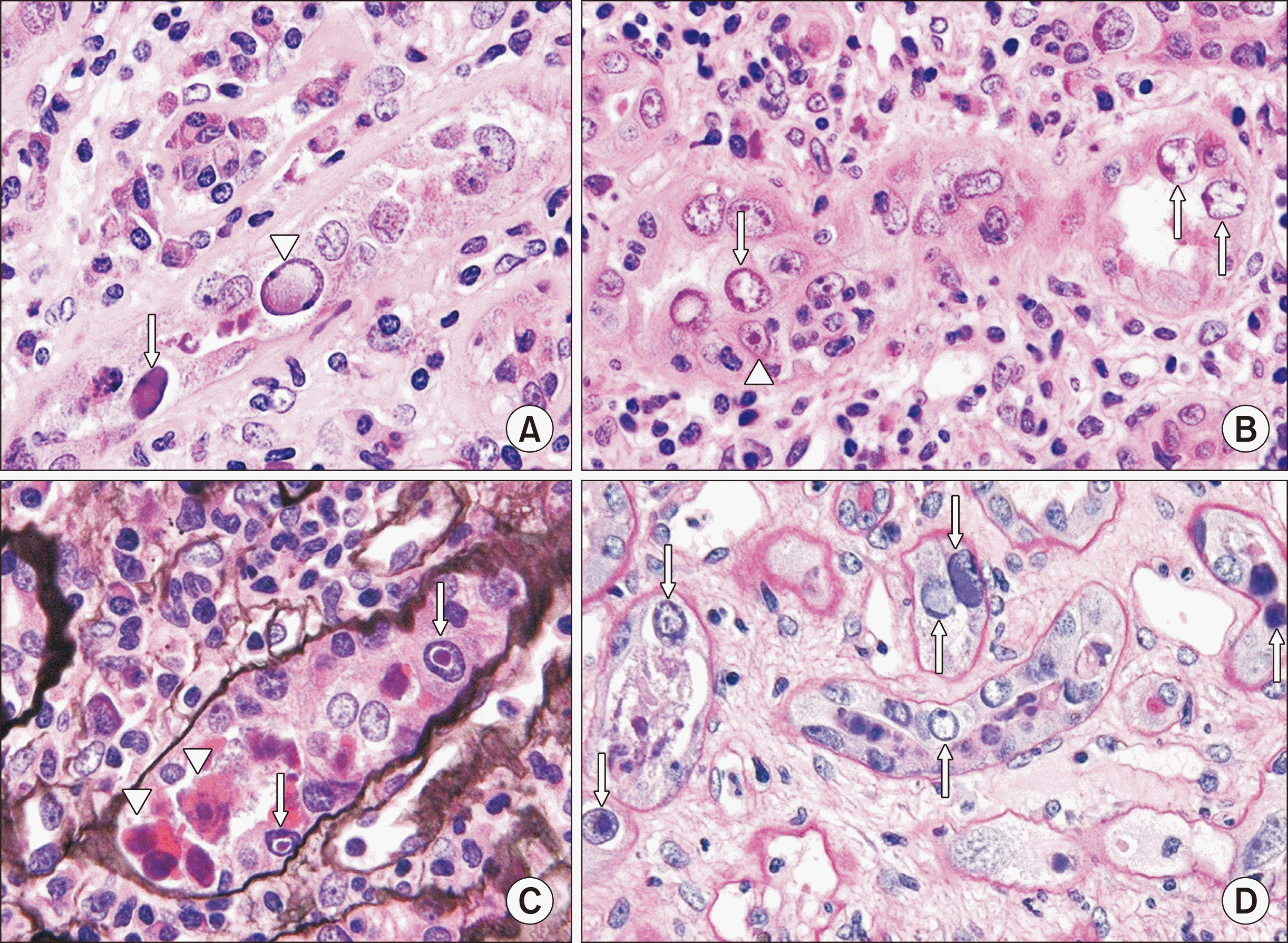
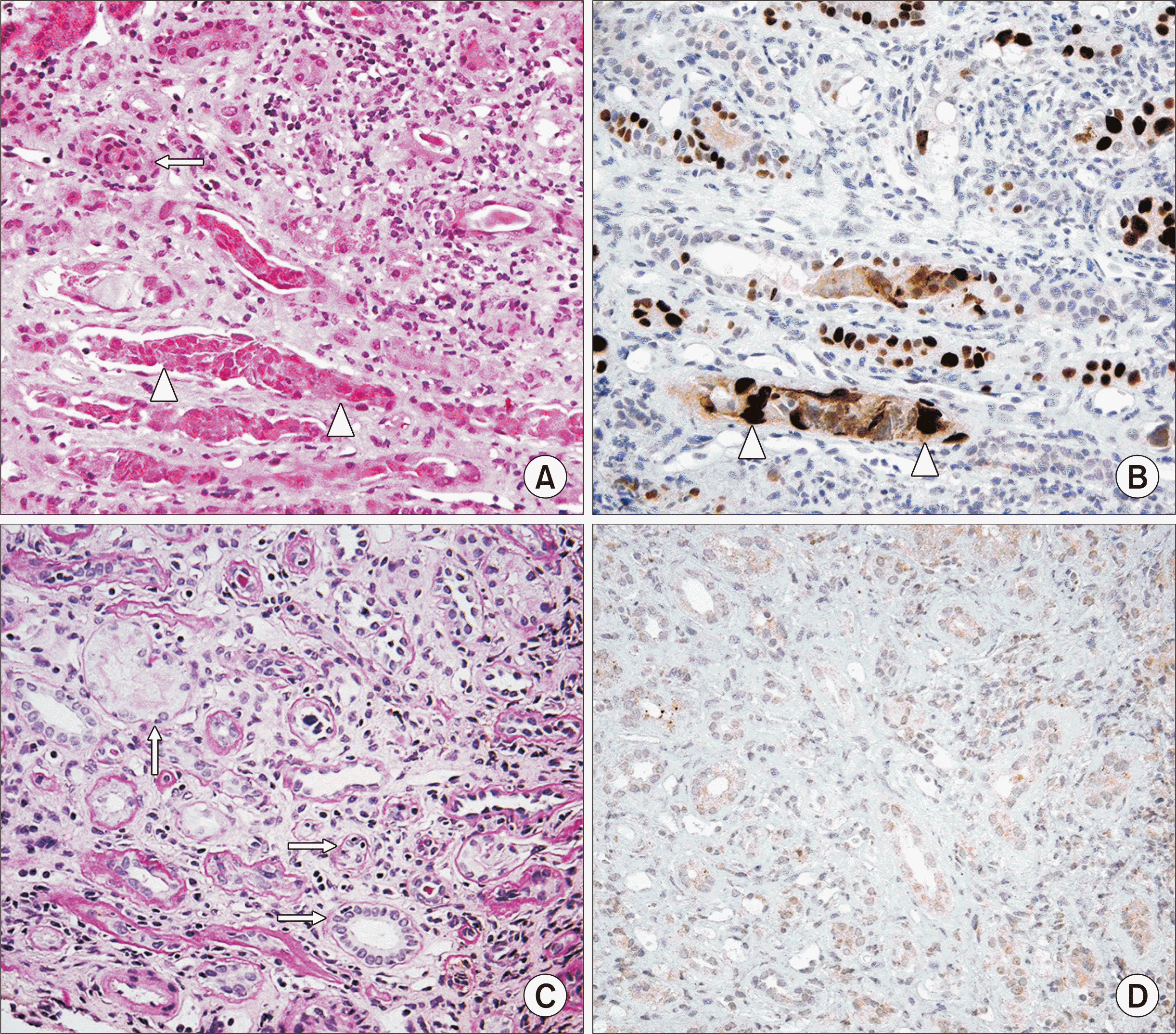
A proven diagnosis of BKVN requires viral identification on kidney biopsy, and several histologic changes are associated with intrarenal PV infection [58]. The virus infects epithelial cell nuclei, favoring distal tubules and collecting ducts. PV infection may be present even when there are no tubular cell abnormalities, although the virus usually induces cytopathic changes, including nuclear enlargement, a "ground glass" appearance or cleared nuclear inclusions, or halos around reddish nuclear inclusions (Fig. 1). On electron microscopy, BK virions are intranuclear nonenveloped particles measuring 30–50 nm in diameter, which may have no organization or occur in paracrystalline arrays (Fig. 2). However, persistent intranuclear dormant virions can exist, so this finding is not diagnostic of active viral replication and BKVN [59]. There is frequent necrosis of tubular cells, which most often show viral inclusions; this is thought to be secondary to virus-mediated injury [60]. Glomerular parietal epithelial cells (PECs) may also be infected. Other histologic features suggestive of PV infection include sharply demarcated geographic interstitial inflammation due to the infection of tubules in a focal manner, eliciting an inflammatory response. There is a mixed inflammatory infiltrate, which may be plasma cell-rich or contain scattered neutrophils, with tubulitis ranging from absent to mild (Fig. 3). In one study of 20 biopsies with PVN, 50% were plasma cell-rich (plasma cells comprising >15% of the inflammatory infiltrate) with a predominance of immunoglobulin M-positive plasma cells, which correlated with high anti-BKV antibody levels [61]. Clusters of macrophages surrounding tubules also may be seen, possibly in response to virally induced tubular damage [62]. While these findings are highly suggestive of PV infection, they are not definitive, because other viruses, such as adenovirus and cytomegalovirus, may induce similar changes. Therefore, it is necessary to identify the inclusions as PV using immunohistochemical staining (IHC) against the SV40 large T antigen (Fig. 4).
A proven diagnosis of PVN requires the identification of viral replication in the kidney, typically by IHC for SV40. In situ hybridization also can be performed, but it is more expensive and less widely available; thus, it is not often used [37,39,41]. Notably, SV40 stains both BKV and JCV; therefore, if SV40 is positive in the setting of negative BK viremia, the infection likely is due to JCV, which should be evaluated serologically. Interestingly, a subset of BKVN kidney biopsies will have tubular basement membrane immune complex deposits containing the BKV antigen, which is associated with viral antigen shedding and more severe tubulitis and fibrosis [63,64]. Therefore, the presence of such immune complex deposits should prompt a search for the underlying cause, including BKVN as well as other disease processes associated with tubular basement membrane deposits [65].
There are some caveats regarding kidney biopsy findings and IHC staining for the diagnosis of BKVN, the most important of which is sampling error due to the focal nature of BKV tubular cell infection [66]. The virus preferentially infects medullary tubular structures and also may be seen in the deep cortex [67]. When both the cortex and medulla are sampled, the virus is identified in up to 58% of cases and is seen only in the medulla in approximately 10% of cases. The medulla also tends to show more extensive tubular cell infection with PV [67]. When only the cortex is sampled, the BKV identification rate may fall to 19%–24%. In up to 40% of biopsies with two tissue cores from the cortex and/or medulla, BKV staining is discordant between the cores [66]. Therefore, at least two cores of kidney tissue should be obtained at biopsy, preferably containing the medulla. BKV inclusions are seen less often in very early and late BKVN, and when the infection has resolved [37]. However, if kidney biopsy IHC for BKV is performed when the plasma viral load is >1,000,000 copies/mL, there is viremia with a >20% reduction in kidney function, there are gene rearrangements of the viral NCCR, or urinary Haufen are present, then the false negative rate has been reported to be <10% [38].
Alternative Approaches to the Diagnosis of BK Virus Nephropathy
Given the reliability issues with BKV plasma and urine quantification, and the focality of kidney biopsy SV40 staining, other approaches for diagnosing BKVN have been explored. Ding et al. [68] tested urinary cell mRNA levels for the PV major capsid structural protein VP1, which participates in viral replication and indicates active BKV transcription, as an independently validated noninvasive biomarker for active intrarenal viral infection and a diagnosis of BKVN. They determined that >650,000 BKV VP1 mRNA copies/ng total urinary RNA had a sensitivity of 93.8% and a specificity of 93.9% for BKVN [68]. In a follow-up study, this level of urinary cell VP1 mRNA was diagnostic of BKVN with a sensitivity of 100% and a specificity of 97% [69]. In addition, concurrently elevated levels of urinary cell mRNA for granzyme B (>11 mRNA copies/mg total RNA) and proteinase inhibitor-9 (>10 mRNA copies/mg total RNA) were associated with loss of graft function in BKVN and were similar to levels in patients with acute rejection. One drawback of this method is that RNA is less stable and less easily isolated than DNA, making DNA testing more reliable and available on a wider basis.
Urinary Haufen have been reported to have high positive and negative predictive values for BKVN. Haufen are dense three-dimensional aggregates of at least six distinct tightly clustered virions that are identified in the urine by negative staining electron microscopy. They form in tubular lumens as cast-like structures from virions released by lysed infected tubular epithelial cells [70,71]. This test is specific for intrarenal BKV infection and the number of Haufen/mL urine tightly correlates with the severity of BKVN [72]. In a study of 809 patients with findings ranging from no evidence of BKV infection to proven BKVN, detection of Haufen had a sensitivity of 100% and a specificity of 98% for BKVN. This outperformed viremia of >10,000 copies/mL, which had a sensitivity of 66% and a specificity of 80% [72]. However, testing for Haufen is not widely available, is time-consuming, is not standardized across centers, and has not received regulatory approval by the U.S. Food and Drug Administration or the European Medicines Agency. Nevertheless, the high predictive value of this test warrants further prospective validation studies. An additional suggested urinary test is double staining of shed epithelial cells with anti-58-kDa Golgi protein, expressed on proximal tubules, and SV40. The presence of urinary 58-kDa+/SV40+ cells is strongly correlated with BKVN, with a positive predictive value of 89.7% (95% confidence interval [CI], 71.5%–97.3%) and a negative predictive value of 91.5% (95% CI, 78.7%–97.2%) [73].
Donor-derived cell-free DNA (dd-cfDNA) has been evaluated as a potential tool for the identification of BKVN. Kant et al. [74] found that dd-cfDNA levels were significantly higher in patients with BKVN than in those with BK viremia and were correlated with the degree of viremia. However, there was overlap with dd-cfDNA levels observed in cell-mediated rejection (CMR). Therefore, this test may suggest progression from viremia to BKVN but cannot exclude rejection, making it of questionable value [74].
Screening for Polyomavirus Nephropathy
Screening for PVN is important to identify disease as early as possible in the hope of preventing graft dysfunction and loss. There are several screening protocols with minor differences, and the implementation varies in different centers, likely reflecting local immunosuppression practices and experience using the detection methods described above [75]. The AST Infectious Disease Community of Practice (AST-IDCOP) has recommended initial monthly screening for plasma BKV DNA for the first 9 months, followed by screening every 3 months until 2 years posttransplantation or if there is graft dysfunction. Alternative screening methods include examining urine for >3 decoy cells/high power field or >1,000,000 DNA copies/mL. If BKV DNA is detected, testing should be repeated to determine if there is an increasing viral load of >1,000 to >10,000 copies/mL plasma, if the latter is sustained for at least 3 weeks, or if urine Haufen are present. If any of these occur and there is no decline in kidney function, it is recommended to reduce immunosuppression with follow-up testing for plasma viral DNA. If there is a reduction in kidney function, then kidney biopsy is recommended to determine the cause [38]. Another suggested screening protocol includes monthly plasma viral DNA testing for 6 months after transplantation, followed by testing every 3 months until 2 years after engraftment and annual testing for an additional 3 years. For patients with >1,000 copies/mL plasma and normal kidney function, immunosuppression is reduced and the viral load is monitored every 2–4 weeks. Kidney biopsy is performed for any unexplained reduction in kidney function or if the elevated serum creatinine and/or viremia do not resolve despite reduced immunosuppression [76]. In pediatric kidney transplant recipients, testing for plasma BKV DNA is recommended every 2 weeks for the first 3 months, followed by monthly testing for 12 months, then testing every 3 months until 3 years posttransplantation. In patients with a high viral load, plasma viral DNA is assessed every 2 weeks. Patients with any viral load undergo kidney biopsy if there is an unexplained rise in the serum creatinine level [77]. These protocols are similar in that plasma BKV DNA is the predominant testing modality used for screening, with immunosuppression reduction and following viral loads are recommended if there is a persistently high viral copy number, and kidney biopsy is recommended for reduction in allograft function. Whenever a kidney biopsy is performed, IHC staining for PV should be performed.
HISTOLOGIC CLASSIFICATIONS OF POLYOMAVIRUS NEPHROPATHY
Two histologic classifications of PVN/BKVN are currently the most widely used; they share some similarities and correlations but also exhibit differences in their approaches [57]. A classification system should encompass the clinical presentation at the time of biopsy and predict the subsequent risk of declining kidney function and graft failure. The histologic features used in these classifications include the extent of tubular cell infection and degree of parenchymal scarring in both, and the severity of interstitial inflammation in one (Table 2). Interestingly, the progression of fibrosis and tubular atrophy has been shown to correlate with the extent of initial inflammation and subsequent tubulitis, independent of the viral load [60]. Therefore, it is recommended to use both classifications for cases of proven BKVN [38].
AST-IDCOP Modified Classification
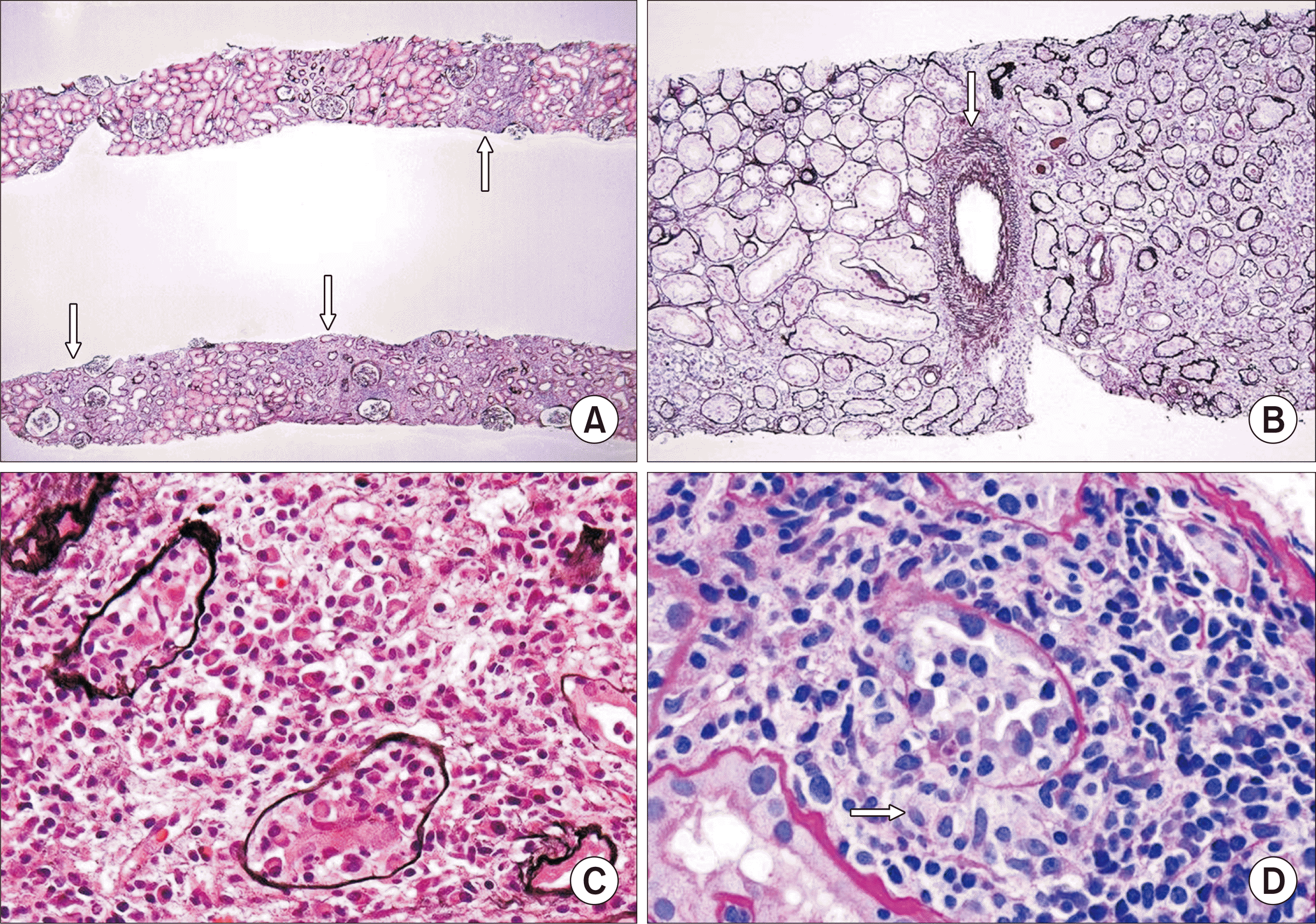
An initial BKVN classification was proposed by Drachenberg at the University of Maryland, subsequently modified with contributions from the AST, and further revised by the AST-IDCOP to include a more detailed description of interstitial inflammation and the extent of tissue involvement by PV [32,39,66,78]. This system broadly classifies PVN based on biopsy findings according to the progressive extent of infected parenchyma characterized by tubular cell viral cytopathic changes with or without positive SV40 staining, interstitial inflammation, and interstitial fibrosis/tubular atrophy (IFTA), which can be characterized by Banff scoring parameters (Table 2, Fig. 5) [79]. Class A encompasses <25% parenchymal involvement with infected tubules with minimal interstitial inflammation and IFTA. Class B includes a spectrum of these features and is subdivided into B1, B2, and B3, which have progressively more widespread viral infection, interstitial inflammation, and IFTA. Class C has severe IFTA, albeit with variable extents of viral infection and interstitial inflammation, because with advancing chronic injury in BKVN, the inflammatory infiltrate may change and the virus may clear. One caveat is that not all biopsies showing BKVN fit into one of the AST-IDCOP classes. For example, a biopsy showing extensive PV infection (>50%) with severe interstitial inflammation (ti3) but no significant tubular atrophy or interstitial fibrosis (IFTA 0) is not classifiable in this system. Class C has been observed more frequently in biopsies performed longer after transplantation [80]. A study by Li et al. [81] demonstrated that with disease progression, kidney biopsies with class C compared to class A had significantly more CD3+, CD4+, and CD8+ lymphocytes, CD128+ plasma cell and CD68+ macrophage infiltrates, and macrophage aggregates.
Banff Classification
A working proposal for a PVN classification was proposed by the Banff group in 2009, initially with classes A, B, and C reflecting the extent of tubular cell viral cytopathic changes and interstitial fibrosis; the degree of interstitial inflammation was not included, as it was not consistently identified during active infection and tubulitis was found to be absent in some infected tubules [82]. The initial classification underwent modifications using statistical analysis with various models and linear regression, resulting in the current Banff classification for definitive PVN. It includes more specifically defined levels of intrarenal PV infection than in the AST-IDCOP classification, and the extent of interstitial fibrosis determined by the Banff “ci” score (Table 2, Fig. 6) [42,83]. There are three classes. Class 1 shows <1% tubules with viral replication and no or mild fibrosis. Similar to the AST-IDCOP class B, class 2 has minor, moderate, or more extensive (>10%) tubular cell infection, with correspondingly moderate to severe, any extent, or minor fibrosis, respectively. Class 3 biopsies have >10% tubules with viral replication and moderate or severe fibrosis. The reproducibility of the early Banff classification system was moderate, with a Κ score of 0.47 [84]. There appears to be a relatively good correlation between Banff PVN class 1 and AST-IDCOP class A, with a variable correlation for classes 2 and B, and a poor correlation for classes 3 and C [62]. Similar to the AST class C, a diagnosis of Banff class 3 has been significantly associated with a longer time from transplantation to kidney biopsy compared to class 1 (P=0.002) and class 2 (P=0.033) [85].
Histologic Classifications and Viral Clearance
The Banff classification has been evaluated with respect to the prediction of viral clearance following treatment by reduction of immunosuppression. Lower kidney biopsy viral loads (polyomavirus load [pvl] 1, pvl 2) have been associated with a higher cumulative rate of BKV clearance, which may be independent of initial BKV plasma load and treatments [57,85]. Cleenders et al. [57] also showed that patients with PVN class 1 had more rapid viral clearance than those with class 2 or 3. Nickeleit et al. [43] reported a significant correlation of PVN class with viral resolution identified by absent kidney biopsy SV40 staining. In addition, PVN classification seems to provide better insight into disease resolution, which is seen in up to 80% in PVN class 1 and 40%–50% in class 3 [7]. In contrast, other studies have not found a significant effect of the Banff PVN class on plasma viral clearance [62,86]. For the AST-IDCOP classification, an early study by Drachenberg et al. [66] found that viral resolution was correlated with a lower histological grade in the index biopsy. However, this was not confirmed in later studies, which failed to find any significant correlation between the BKVN classification and viral clearance or the extent of interstitial inflammation [57,62].
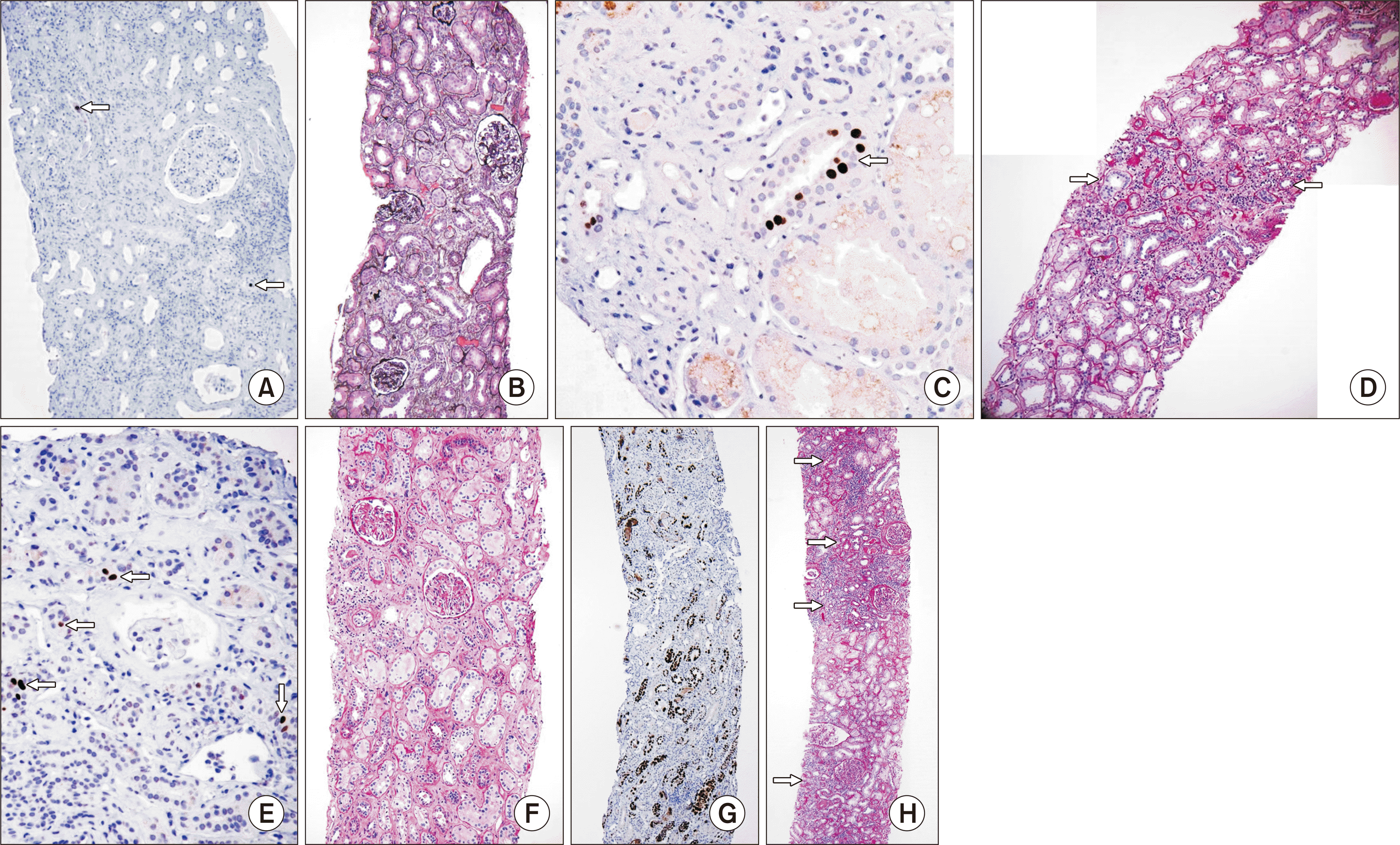
Viral clearance may alter the histologic appearance of BKVN after the reduction of immunosuppression (Fig. 7). As the virus clears, a form of immune reconstitution has been reported in the first 2 months of treatment, leading to an increase in the severity of tubular and interstitial inflammation, the number of inflamed tubules, and the extent of interstitial plasma cells [62,87–89]. These changes occur in concert with a reduction of tubular cell SV40 staining, typically without an increase in interstitial fibrosis or tubular atrophy. These biopsy changes likely reflect an augmented antiviral response, but may mimic CMR, potentially making biopsy interpretation difficult. The median time to plasma viral clearance is 9 months, but a repeated biopsy 4 months after BKVN treatment initiation has been shown to be reflective of virally induced kidney damage and predictive of the long-term prognosis [87].
Histologic Classifications and Clinical Outcomes
Several studies have evaluated kidney function and allograft loss with respect to BKVN histologic classifications, with mixed results. With respect to the Banff classification, in an initial study Nickeleit and Singh [7] demonstrated that PVN class 1 had a 2-year graft survival rate of 90%, versus 70% for class 2 and 50% for class 3. This correlation between Banff PVN classes and outcomes was corroborated in a subsequent study that showed a significant difference in serum creatinine levels among all Banff PVN classes, with the largest difference found between classes 1 and 3 (P=0.001). Similarly, at 24 months, that study demonstrated a significant difference with respect to graft failure (class 1, 16%; class 2, 31%; class 3, 50%; P=0.004); both parameters showed this relationship irrespective of rejection or comorbid conditions [84]. An additional study found significant loss of kidney function at 24 months only in class 3 compared to classes 1 and 2, but graft loss was significantly different among all PVN classes when evaluated more than 24 months after diagnosis (class 1, 5%; class 2, 30%, class 3, 50%; P=0.009) [43]. A smaller study also demonstrated an association of the PV load and PVN class with subsequent graft function, although concurrent rejection may have been a complicating factor [90].
In contrast, several investigators have failed to find any significant predictive value of the Banff PVN classification with respect to graft failure. A Dutch study found that, while class 3 patients had the worst outcome, the Banff PVN classes showed no significant associations with the trajectory of kidney functional decline (P>0.09) or with death-censored loss of kidney function (P=0.51) at 2 or 4 years of follow-up [91]. Kowalewska et al. [85] found that while patients with class 3 had significantly worse serum creatinine levels compared to classes 1 (P=0.025) and 2 (P=0.037), there was no significant difference in the rate of graft failure at 2 years after biopsy. Other studies also did not find any statistical associations between Banff PVN classes and graft loss [62,86].
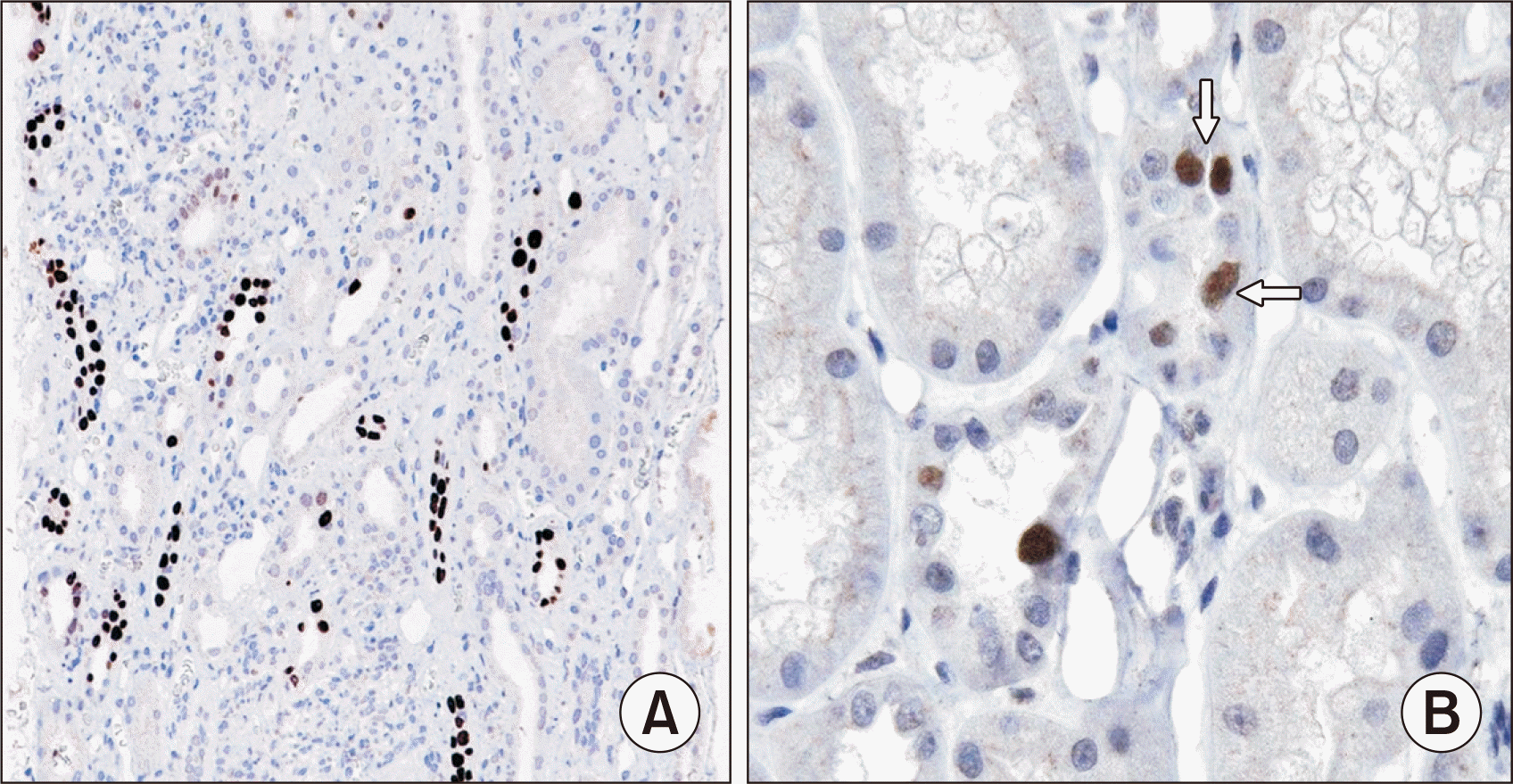
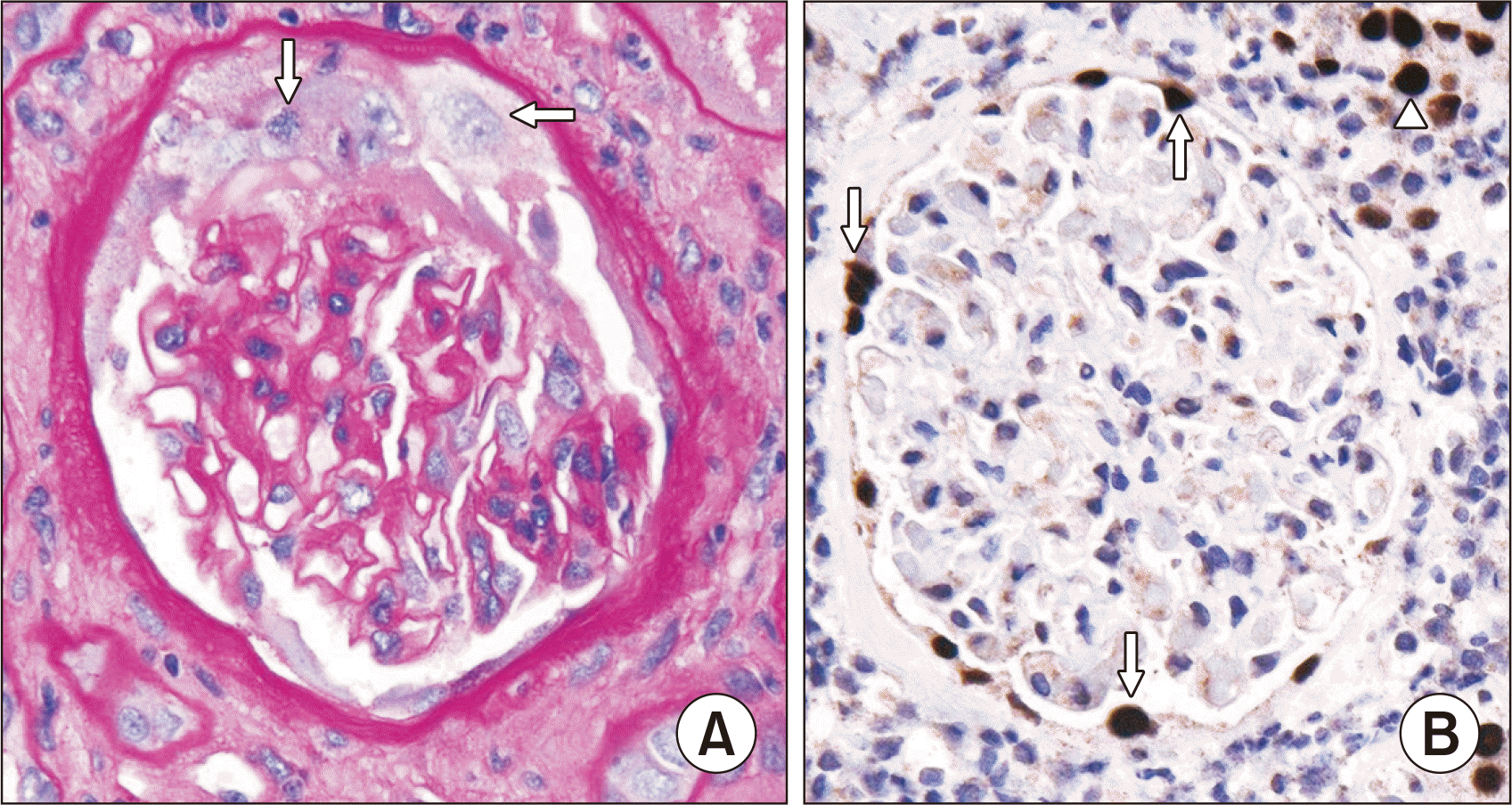
Fewer studies have assessed the value of the AST PVN classification in predicting kidney functional outcomes. Drachenberg et al. [66] evaluated an earlier iteration of the AST classification and found a significant correlation (P=0.0002) of all PVN classes with graft loss over at least 12 months postbiopsy. However, graft loss also correlated with the serum creatinine level at the time of biopsy, and there were small patient numbers for classes B3 and C [66]. Other studies showed increased graft loss in those with PVN class C than in classes A and B on the initial kidney biopsy (P<0.05) [80,83]. However, a later study by Drachenberg et al. [62] did not support the use of arbitrary histologic quantitative features or classifications in predicting graft outcomes. One additional histologic feature suggested to correlate with graft outcomes is BKV infection of glomerular PECs (Fig. 8). Patients with infected PECs had more advanced BKVN based on the AST classification system (P<0.01) than those with uninfected PECs, with more tubulointerstitial inflammation and scarring, higher serum creatinine levels at follow-up (P=0.003), and reduced death-censored graft survival at 125 months after initial biopsy (P=0.004) [92]. Thus, the presence of infected PECs in BKVN appears to be associated with more severe infection and to be a harbinger of increased risk of graft loss.
DIFFERENTIAL DIAGNOSIS OF TRANSPLANT REJECTION AND POLYOMAVIRUS NEPHROPATHY
Transplant rejection may occur preceding, concurrent with, or following BKVN, and it may be difficult to differentiate these disparate forms of transplant kidney injury. Acute rejection has been reported in up to 38% of patients during or following BKVN, with most studies reporting up to 15% and with subclinical rejection occurring in another 20%–33% [31,55,62,93,94]. This typically follows, and likely is secondary to, a reduction in immunosuppression as an antiviral measure, although it can occur at the time of initial BKVN diagnosis [7]. Other significant risk factors for acute rejection include transient or persistently high viremia levels [49]. In a study by Gras et al. [15], acute rejection was found in 13.8% of patients with BKVN at a median of 16 months after infection compared with 3% of matched controls (P=0.003), with a significantly higher level of donor-specific antibodies (DSA) in the BKVN patients. Other investigators also have noted a higher level of DSA following BKVN, and up to 45% of rejection episodes are antibody-mediated or mixed rejection [62,95–98].
BV Virus Nephropathy Versus Cell-Mediated Rejection
There is considerable overlap in the histologic features of BKVN and CMR; however, some features may be helpful in distinguishing these two entities (Table 3). The presence of tubular epithelial cell cytopathic changes and viral inclusions, very geographic inflammation that is often more medullary, a greater number of plasma cells and neutrophils, and tubulocentric macrophage clusters are more suggestive of BKVN. In contrast, acute rejection tends to be characterized by diffuse and more severe tubulitis, without BKV inclusions and with more eosinophils [62,99]. Arterial inflammation is considered diagnostic for rejection with or without coincident viral inclusions and may have a worse outcome in a background of reduced immunosuppression [100]. It has been suggested that tubulitis more than a ×20 field away from any viral inclusions indicates rejection when CMR and BKVN coexist. Tubular cell staining for major histocompatibility complex class II (HLA-DR) has been suggested as a means for distinguishing BKVN from CMR; however, some studies have found no difference in this finding between CMR and BKVN [39,88,101]. Therefore, further studies are needed to validate its diagnostic use. Elevated levels of dd-cfDNA have also been studied as a means of differentiating BKVN from rejection with higher levels seen in the urine in BKVN; however, this method does not show sufficient discrimination to be useful in this regard [102,103]. Molecular diagnostics have been applied to this dilemma. Diagnostic gene sets from kidney biopsies and from the urine may be of ancillary help in making a diagnosis of BKVN, but to date have not been able to consistently differentiate BKVN from rejection [104–106]. This is a promising area where more studies are needed.
Immunophenotyping of graft-infiltrating and urinary cells has been evaluated as a means to distinguish rejection from BKVN with variable results. Kim et al. [107] used a multiplex immunofluorescence assay to assess cellular infiltrates in different kidney biopsy locations. They found that cortical CD8+ cells were more frequent in CMR (P=0.034), while in BKVN there were more medullary CD20+ cells (P=0.028). In contrast, an Australian study found that the numbers and clusters of C20+ cells and granzyme B staining did not differentiate between CMR and BKVN, but perforin-positive interstitial cytotoxic T cells were more often seen in CMR (P<0.0001) [108]. However, the intrarenal location of the CD20+ cells was not specified in that study. Others have identified more CD20+ cells (P<0.001) and a trend for there to be more CD68+ cells (P=0.055) in kidney biopsies of BKVN versus CMR, with no differences in absolute T cell numbers. However, a higher percentage of the inflammatory cells were T cells in CMR [101]. In a study evaluating urinary and kidney biopsy samples, patients with CMR had significantly higher total numbers of urinary CD8+, CD8+ effector memory and CD8+ terminally differentiated cells than BKVN patients. Kidney biopsies from patients with CMR had more CD4+ (P=0.001) and CD8+ (P=0.004) cells, and a positive correlation was found between the number of CD4+ and CD8+ cells in the renal biopsies and in the urine [109]. Taken together, the data suggest that CMR is associated with more T cell infiltrates, while BKVN has more CD20+ cells, particularly in the medulla, and possibly more CD68+ cells. However, at best these are suggestive findings and are not sufficient to make a differential diagnosis between these forms of kidney injury.
BK Virus Nephropathy Versus Antibody-Mediated Rejection
The diagnostic features of antibody-mediated rejection, including glomerulitis, transplant glomerulopathy, vascular inflammation and fibrinoid necrosis, peritubular capillary basement membrane multilayering, and peritubular capillary C4d staining, are not found in BKVN and usually can distinguish between these entities (Table 3). These features are also helpful in diagnosing antibody-mediated rejection in the setting of concomitant BKVN [62,95,110,111]. In the setting of active interstitial inflammation, peritubular capillaritis likely is of minimal diagnostic use. As noted above, DSA may increase following BKVN, potentiating the risk for antibody-mediated rejection.
CONCLUSION
In summary, BKVN can be diagnosed presumptively by finding a viral DNA copy number of >10,000/mL plasma and proven by identifying infected tubular cells in an allograft biopsy, with the caveat that the virus is found focally in the kidney and may not be present in a tissue sample. There are two commonly used BKVN histologic classifications, both of which correlate with clinical outcomes to some extent. Repeat kidney biopsy at 4 months after the diagnosis of BKVN with evaluation in the context of viral clearance or disease persistence is useful for prognostication. Differentiating CMR in the setting of concurrent or recently treated BKVN is difficult, although there are some suggestive morphologic, immunologic, and molecular findings to assist in this dilemma. However, work remains to be done to develop methods for definitively distinguishing CMR and BKVN, and to ensure appropriate therapy and optimal outcomes for kidney transplant recipients who contract this challenging viral infection.


 XML Download
XML Download