

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Morphine and other opioid medications are widely recognized as effective analgesics for managing moderate-to-severe pain. On the other hand, the development of tolerance to the pain-relieving properties of morphine becomes a notable concern when it is used over an extended period in clinical pain management. Unfortunately, there are currently no effective measures for preventing or treating this phenomenon [1]. The significant impact of morphine tolerance on the effectiveness of pain management underscores the need to comprehend the underlying mechanisms of this phenomenon and discover potential solutions to overcome it, which would have valuable clinical implications. The underlying mechanisms of morphine tolerance are complicated and multifaceted. The complete complement of neurobiological mechanisms underlying morphine tolerance is still unknown, despite significant advancements over the last few years [2,3]. Based on research involving both humans and experimental animals, several causes have been proposed, including apoptosis, oxidative stress, neuroinflammation, and loss or malfunction of the μ-opioid receptor. One of the most probable pathways for the progression of morphine antinociception tolerance is oxidative stress damage [4]. Lipid peroxidation and excessive reactive oxygen species (ROS) generation disrupt redox balance and harm the central nervous system (CNS) [5]. Thus, induced lipid peroxidation, like iron-dependent lipid oxidation, should be regarded in terms of morphine tolerance. Numerous biological functions, such as oxygen transportation and neurotransmitter synthesis, depend on iron. Free-iron overload, on the other hand, results in CNS damage by inducing ferroptosis, which is linked to neurodegenerative illness, and aging [6,7].
Ferroptosis, distinguished from apoptosis, necrosis, and autophagy, is a condition of cell death that primarily relies on oxidative stress and the involvement of iron [8]. The occurrence of ferroptosis is a consequence of the disturbance in cellular antioxidant defenses that rely on glutathione (GSH). This disruption leads to the build-up of detrimental lipid-derived ROS, ultimately resulting in the demise of cells through ferroptosis [9]. Emerging investigation indicates that ferroptosis is complicated in the pathological cell death observed in brain tissues exposed to elevated levels of glutamate (Glu), as well as in kidney and heart tissues experiencing ischemia-reperfusion injury [9,10]. According to a recent study, ferroptosis may be related to prolonged morphine treatment that results in morphine antinociception tolerance. It was shown that morphine tolerance caused increased iron levels in the spinal cord in mice. The ferroptosis inhibitor regulated transferrin receptor protein 1/ferroportin, resulting in attenuation of iron overload and slowing the formation of morphine tolerance by raising GSH peroxidase 4 levels [11]. Recently discovered as a regulatory process of cellular demise, ferroptosis is characterized by distinct morphological features, including the presence of unusually small mitochondria, diminished mitochondrial cristae, and impairment of the external mitochondrial membrane [12]. Countless examinations have shown that oxidative stress can cause ferroptosis in various experimental settings [13]. Interestingly, ferroptosis might be successfully inhibited in vivo by improving antioxidant function [14].
Ferrostatin-1, a ferroptosis inhibitor, significantly more effectively prevents ferroptosis when compared to phenolic antioxidants. Ferrostatin-1 can also suppress iron-induced peroxidation in liposomes [15]. Ye et al. [16] directed an investigation that revealed that the introduction of ferrostatin-1 led to a decrease in cognitive dysfunction among rats with epilepsy. In addition, Chu et al. [17] discovered that the application of ferrostatin-1 offered protection against oxidative toxicity in HT-22 cell lines.
Based on these studies, it is clear that chronic morphine exposure leads to ferroptosis, which causes morphine antinociception tolerance. Thus, it is reasonable to expect that inhibition of ferroptosis would reduce morphine tolerance. Additionally, while the positive impact of ferrostatin-1, an inhibitor of ferroptosis, has been observed in certain CNS disorders, its potential effects on morphine tolerance remain unexplored. Therefore, the main purpose of this investigation is to examine the influence of ferrostatin-1 on the progression of morphine tolerance and comprehend the underlying mechanisms involved in this process in male rats.
MATERIALS AND METHODS
1. Animals
This study involved conducting experiments on 36 adult male Wistar albino male rats, which had an average weight of 220–260 g. The animals were obtained from Sivas Cumhuriyet University Experimental Animal Unit. The rats were kept in polyacrylic cages measuring 38 × 23 × 10 cm, with four animals housed in each cage. They were housed in controlled environmental conditions, maintaining a consistent temperature of 23°C ± 1°C, relative humidity between 54% ± 10%, and a regular light-dark cycle of 12 hours each. Throughout the experiment, the rats had unrestricted access to a commercial rat pellet diet and water. Each experimental group contained six rats. The group size was determined by calculating power analysis. Furthermore, each experimental group in previous studies within this field consisted of six rats [18]. The animals were acclimated to the laboratory circumstances before the experiments took place. All experimental procedures were conducted blindly within a specified time frame of 10 to 15 hours. In the present research, the choice was made to euthanize the animals using the method of cervical dislocation under anesthesia (ketamine at a dose of 90 mg/kg intraperitoneally [i.p.]). Experiment protocols were approved by Cumhuriyet University Animal Ethics Committee on September 22, 2021 (Ethic no: 65202830-050.04.04-590). The experiment protocols also adhered to the Guide for the Care and Use of Laboratory Animals, 8th Edition published by the National Academies Press (US), 2011, and the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
2. Drugs
For the experimental trials, fresh preparations of ferrostatin-1 (obtained from the Sigma-Aldrich Co.) dissolved in dimethyl sulfoxide (DMSO) and morphine sulfate (sourced from Sivas Cumhuriyet University Hospital in Turkey) dissolved in a saline solution were prepared. Before the analgesia tests, morphine (5 mg/kg) [18] and ferrostatin-1 (1 mg/kg) were given subcutaneously (s.c.) and i.p. respectively. While using different routes of administration can introduce a difference in pharmacokinetics, measures were taken to ensure that the chosen routes and doses for each drug were well-established and in line with previous studies [18,19], to facilitate a meaningful comparison. A pilot study was conducted to determine the appropriate dose of ferrostatin-1. Various doses were administered to rats and the dose that effectively reduced morphine tolerance while avoiding toxic effects on the rats was selected.
3. Experimental protocols
The tail-flick test (May TF 0703 Tail-flick Unit; Commat) and hot-plate test (May AHP 0603 Analgesic Hot-plate; Commat) were employed to evaluate the impact of ferrostatin-1 on both morphine analgesia and tolerance. The rats were habituated to the test situations to minimize stress. This was achieved by restraining the rats in the same manner as for the tail-flick and hot plate tests one day before the start of the investigation, but without actually conducting the tail-flick and hot plate tests. The response time was measured at four different time points: 30, 60, 90, and 120 minutes after the administration of the drugs. The rats were divided into six groups for the study: DMSO + saline, 1 mg/kg ferrostatin-1 + saline, DMSO + 5 mg/kg morphine, ferrostatin-1 + morphine, DMSO + morphine tolerance, and ferrostatin-1 + morphine tolerance groups (Table 1). DMSO and ferrostatin-1 were administered i.p., while morphine and saline were administered s.c., all at a volume of 1 mL/kg as specified. After conducting the analgesic tests, the animals were euthanized by cervical dislocation. In this research, the authors chose to euthanize the animals using the method of cervical dislocation under anesthesia (ketamine at dose of 90 mg/kg i.p.). The tissue samples from the dorsal root ganglia (DRG) were then collected from the animals at the T12-L5 levels for further assessment (Fig. 1).
4. Antinociception tests
The evaluation of thermal pain was carried out using tail flick and hot plate tests. In the tail flick test, a radiant heat source was applied to a specific 3 cm section of the rats' tails following the administration of either control or test drugs. The tail-flick latencies were then recorded after the application of radiant heat. To avoid tissue harm, a 15-second cutoff time was chosen. Rats that did not respond after 15 seconds were removed from the experiment. The nociceptive response observed in the tail flick method is primarily attributed to the involvement of spinal mechanisms [20,21]. During the hot plate procedure, the rats were positioned on a heated plate set at a precise temperature of 53°C ± 0.5°C. The time it took before the animal began to lick its paws or jump to get away from the heat was noted as a pain threshold indicator. To avoid damaging, a 30-second cutoff time was chosen. Both spinal and supraspinal mechanisms contribute to the nociceptive response observed in the hot plate method [21]. The primary reason for utilizing both tail flick and hot plate tests was to ensure a comprehensive evaluation of thermal nociception in the experimental model. Each of these tests provides unique insights into thermal pain responses, and their combination allows for a more robust assessment of the drug's impact on thermal hypersensitivity. The tail flick test primarily evaluates the spinal reflex withdrawal response to a noxious thermal stimulus, while the hot plate test assesses the response to a more prolonged thermal stimulus involving both spinal and supraspinal mechanisms [22]. By employing both tests, the authors aimed to capture different aspects of thermal nociceptive processing, providing a more complete picture of the drug's effects on thermal pain perception.
5. Morphine tolerance induction
Rats were randomly selected to form the morphine tolerance groups. To generate morphine tolerance, the rats received subcutaneous injections of 10 mg/kg of morphine twice daily (at 10:00 and 17:00) for a period of five consecutive days. In order to evaluate the influence of ferrostatin-1 at a dose of 1 mg/kg on morphine tolerance, the drug was administered 30 minutes prior to the administration of morphine for a period of five days. The groups treated with ferrostatin-1 alone received a control injection 30 minutes after ferrostatin-1. The groups treated with morphine alone received a control injection 30 minutes before the morphine. On the sixth day, the rats were administered the optimal analgesic dose of morphine, which was 5 mg/kg, via subcutaneous injection. Subsequently, the analgesia tests were conducted at 30-minute intervals (30, 60, 90, and 120 minutes) to assess the level of tolerance developed. The animal model used for morphine tolerance induction was previously applied in studies such as the one conducted by Taskiran et al. [23]. Twelve rats were utilized to create a morphine tolerance model, with 6 of them receiving ferrostatin-1 in conjunction with morphine, as detailed in the table above. It was determined that rats became the model for morphine tolerance when the tail-flick and hot plate latencies, representing the time taken by the rats to respond to thermal stimuli, significantly decreased in rats receiving repeated doses of morphine compared to those receiving a single dose of morphine.
6. DRG tissue homogenate preparation
After collecting the DRG tissue samples from the rats, they were immersed in cold phosphate-buffered saline. The samples were then homogenized using a mechanical homogenizer (SpeedMill PLUS; Analytik Jena) in a chilled phosphate buffer saline solution with a pH of 7.4. Following homogenization, the samples were subjected to centrifugation at 4,000 rpm for 10 minutes at a temperature of 4°C. The resulting supernatants were carefully collected and stored at –80°C until further biochemical analysis. The total protein levels in the samples were quantified using a Bradford protein assay kit (Merck) [24].
7. Determination of GSH, glutathione peroxidase 4 (GPX4), and nuclear factor erythroid 2-related factor 2 (Nrf2) levels
The levels of GSH, GPX4, and Nrf2 in the supernatants of the DRG were determined using commercially available rat ELISA kits (YL Biont). The experimental procedures followed the instructions provided by the manufacturer. In the experimental procedure, both standard and tissue samples were placed in a plate and incubated for 60 minutes at a temperature of 37°C. After the washing step, staining solutions were added and allowed to incubate at 37°C for 15 minutes. A stop solution was introduced, and the absorbance of each sample was measured at 450 nm. To quantify the samples, standard curves were constructed. The coefficients of variation within and between plates were below 10%, indicating good precision and reproducibility.
8. Determination of total antioxidant status (TAS) and total oxidant status (TOS) in the DRG tissue
The analysis of TAS and TOS levels in the DRG tissue was conducted using an automated assay method specifically developed by Erel [25,26]. To measure TAS, the reaction rate of free radicals was monitored by evaluating the absorbance of colored dianisidyl radicals that are formed during free radical reactions. This measurement process initiates with the generation of hydroxyl radicals in a Fenton reaction. The color intensity observed in the tissue samples is anticipated to decrease in proportion to the antioxidant content present. Conversely, TOS measurement involves the oxidation of ferrous ions to ferric ions in the presence of sufficient oxidants. This technique allows for the determination of TOS levels by quantifying the concentration of ferric ions in the tissue using xylenol orange. This assay was calibrated using hydrogen peroxide. TAS and TOS kits used in the measurement were obtained from Rel Assay Diagnostics, Turkey. The experimental procedures were conducted following the instructions provided by the manufacturer. The data were represented as μmol Trolox Eq/g protein for TAS and μmol H2O2 Eq/g protein for TOS [27].
9. Data analysis and statistical test
The calculation of the percentage of the maximal anti-nociceptive effects (% MPE) involved applying the following formula:
% MPE = [(test latency – baseline) / (cutoff (30) – baseline)] * 100 [28].
The obtained findings were presented as mean ± standard error of the mean. Normal distribution was evaluated using the Shapiro–Wilk test. The data were analyzed using one-way analysis of variance (ANOVA), with multiple comparisons assessed using the Tukey test. SPSS software (version 22.0; IBM Co.) was utilized for these analyses. Statistical significance was established for all groups with a significance level set at P < 0.05.
RESULTS
1. The impact of ferrostatin-1 on nociception and morphine analgesia
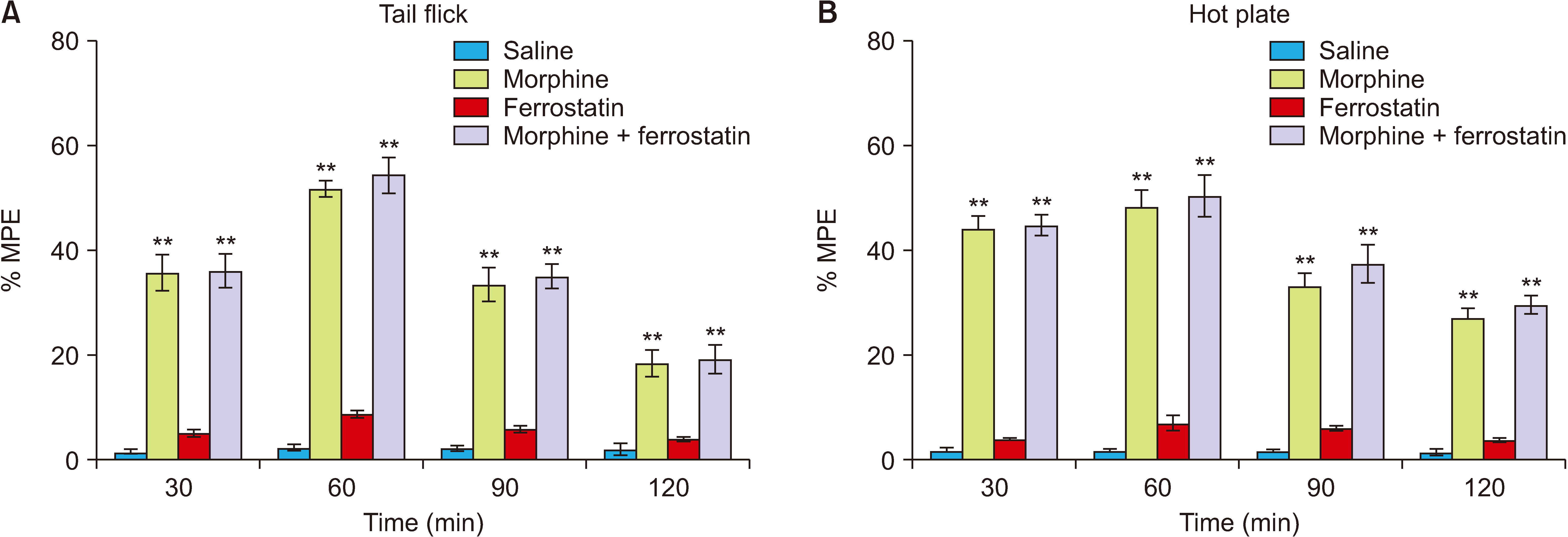
The analgesia tests were conducted over 120 minutes at 30-minute intervals to assess the influence of ferrostatin-1 on nociception and morphine analgesia at a dose of 1 mg/kg. In comparison to the control group, ferrostatin-1 did not exhibit any significant antinociceptive effect in either the tail flick or hot plate tests (P > 0.05; Fig. 2). Similarly, when compared to the morphine group, ferrostatin-1 did not alter the antinociceptive effects of morphine in either the tail flick or hot plate tests (P > 0.05; Fig. 2).
2. The impact of ferrostatin-1 on the development of morphine tolerance
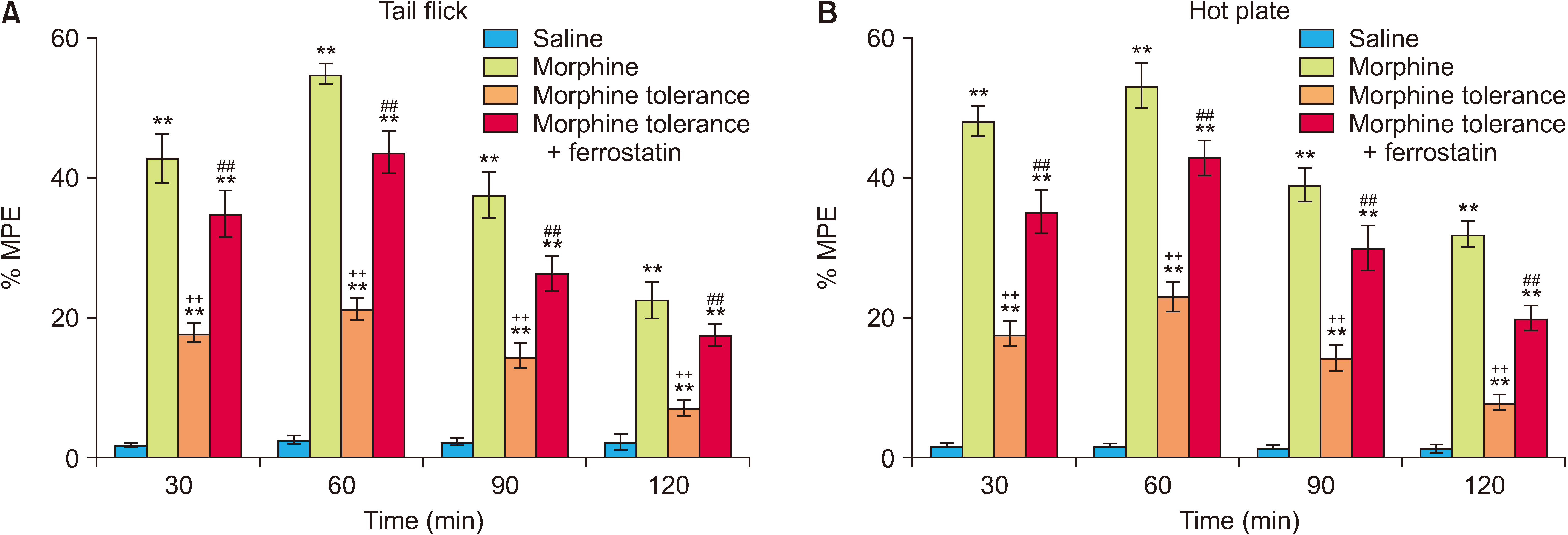
At all the time points, the % MPE values obtained from the tail flick and hot plate tests were consistently higher in the morphine group compared to the morphine tolerance group, indicating a significant difference (P < 0.001; Fig. 3). The co-administration of ferrostatin-1 with the induction of morphine tolerance resulted in a notable decrease in the development of morphine tolerance in both the tail flick and hot plate tests at all examined time points (P < 0.001; Fig. 3). The results of the ANOVA for the tail flick and hot plate tests are presented in Tables 2 and 3, respectively. These tables display the statistical outcomes, including F values, degrees of freedom, and P values, for the comparisons among experimental groups in each test.
3. The impact of ferrostatin-1 on GSH, GPX4, and Nrf2 levels in morphine analgesia and tolerance in the DRG
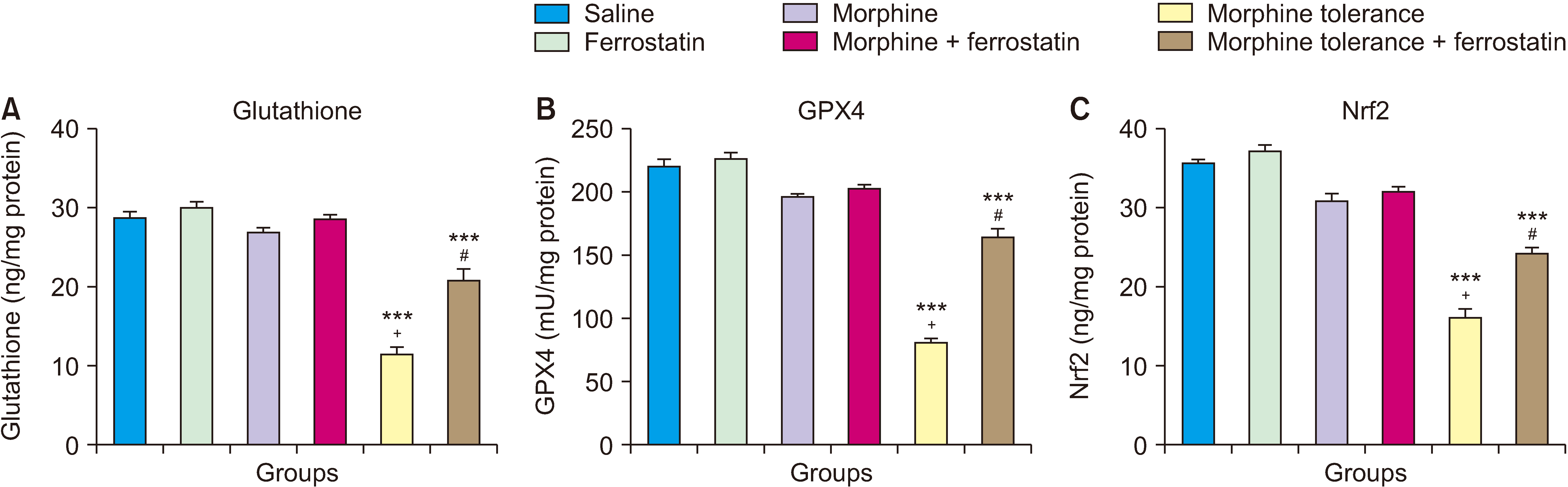
A single dose of morphine did not significantly impact the levels of GSH, GPX4, and Nrf2 in the DRG compared to the control group (P > 0.05; Fig. 4). However, the development of morphine tolerance led to a decrease in the levels of GSH, GPX4, and Nrf2 in the DRG compared to both the control group (P < 0.001; Fig. 4) and the group treated with a single dose of morphine (P < 0.001; Fig. 4). On the other hand, when ferrostatin-1 was administered along with morphine tolerance, it increased the levels of GSH, GPX4, and Nrf2 in the DRG compared to the morphine tolerance group (P < 0.001; Fig. 4). The results of the ANOVA for the ELISA tests of GSH, GPX4, and Nrf2 are presented in Table 4. This table displays the statistical outcomes, including F values, degrees of freedom, and P values, for the comparisons among experimental groups in each test.
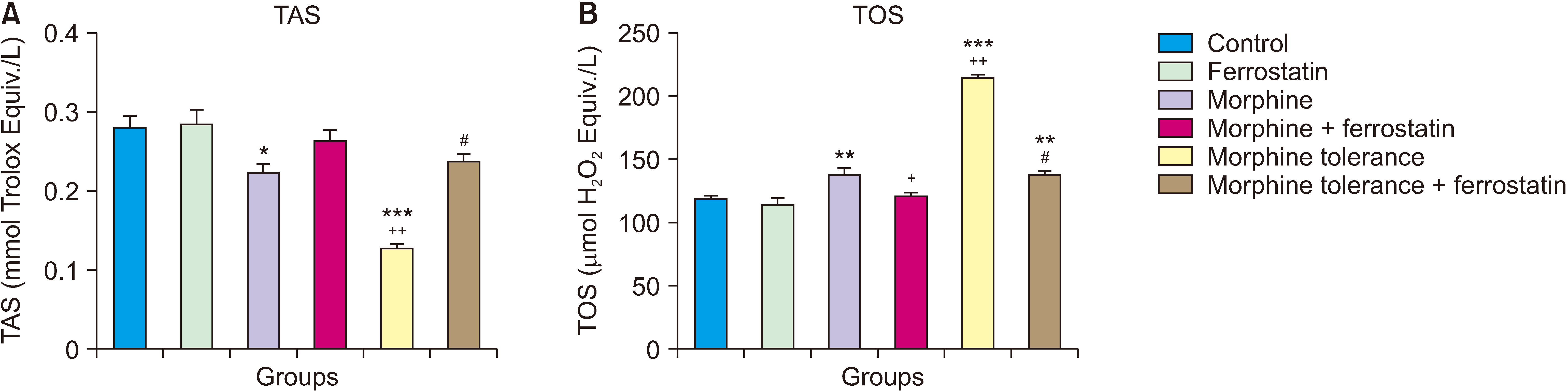
4. The effect of ferrostatin-1 on TAS and TOS levels in morphine analgesia and tolerance in DRG
In comparison to the control group, a single administration of morphine resulted in a significant decrease in TAS levels (P = 0.017; Fig. 5A) and a significant increase in TOS levels (P = 0.001; Fig. 5B) in the DRG. Compared to the group that received a single dose of morphine, the addition of ferrostatin-1 in the treatment significantly decreased TOS levels (P = 0.006; Fig. 5B) in the DRG when combined with morphine. In comparison to the control group and the group treated with a single dose of morphine, morphine tolerance led to a significant decrease in TAS levels and an increase in TOS levels in the DRG (P < 0.001; Fig. 5). However, in the presence of tolerance development, the addition of ferrostatin-1 resulted in a significant increase in TAS levels and a decrease in TOS levels in the DRG compared to the group with morphine tolerance (P < 0.001; Fig. 5). The results of the ANOVA for the TAS and TOS tests are presented in Table 5.
DISCUSSION
Ferroptosis, recently discovered, is a distinctive type of regulated cell death characterized by the accumulation of lipid peroxides. This buildup is triggered by the disruption of antioxidant mechanisms reliant on GSH. It distinguishes itself from apoptosis, autophagy, and necrosis through genetic, morphological, and biological differences [29]. Ferroptosis initiates lipid peroxidation through the production of ROS via the Fenton reaction [30]. New research reveals that ferroptosis plays a role in drug resistance [31]; for example, the lipid peroxidation pathway is implicated in anticancer drug resistance. Other features of ferroptosis include the development of oxidative bursts, depletion of antioxidants, and stimulation of lipid peroxidation [32]. Previous research has demonstrated that ferroptosis is crucial in neurological diseases [33]. It was shown that ferroptosis, characterized by free-iron overload, contributes to morphine tolerance [11]. The use of ferrostatin-1, which is an inhibitor of ferroptosis, has been associated with positive outcomes in the CNS. Li et al. [19] demonstrated that ferrostatin-1 attenuated mechanical hypersensitivity by suppressing spinal ferroptosis in rats with streptozotocin-induced diabetes. An et al. [34] indicated that ferrostatin-1 reversed acrylamide-induced biological activities and facilitated the repair of damaged DRG neurons by inhibiting ferroptosis.
Li et al. [35] revealed that injections of ferrostatin-1 into the striatum and cerebral ventricles produced neuroprotective effects after collagenase-induced intracerebral hemorrhage. In the autologous blood infusion model of intracerebral hemorrhage, intraperitoneal injection of ferrostatin-1 was demonstrated to enhance long-term neurological processes [36]. The positive effects of ferrostatin-1 on the CNS, coupled with the hypothesis that inhibition of ferroptosis may reduce morphine tolerance, prompted us to investigate the impact of ferrostatin-1, a ferroptosis inhibitor, on morphine tolerance, a topic not previously studied. The primary objective of this study was to examine the impact of ferrostatin-1 on acute pain, morphine analgesia, and the development of morphine tolerance, along with investigating the underlying mechanisms responsible for these effects in male rats. The DRG are crucial in transmitting sensory information from peripheral tissues to the CNS [37]. Given that opioids, including morphine, primarily act on the peripheral nervous system to alleviate pain, the authors believed that investigating molecular changes in the DRG would provide valuable insights into the early events that contribute to morphine tolerance. In the present work, ferrostatin-1 did not exhibit any analgesic effects and had no impact on the analgesic effect of single-dose morphine. However, it effectively reduced the development of tolerance to repeated doses of morphine. These outcomes are consistent with an earlier investigation conducted by Chen, which reported that suppression of ferroptosis by liproxstatin-1 suppressed tolerance development, while activation of ferroptosis by erastin accelerated it [11]. Chen et al. [11] also discovered that chronic morphine treatment caused neuronal loss, iron accumulation, inflammation, lipid peroxidation, and mitochondrial shrinkage in the spinal cord, but liproxstatin-1, a ferroptosis inhibitor, reversed all of these events.
The hot plate and tail flick tests are both experimental methods used in laboratory settings to assess pain sensitivity and nociceptive responses in animals, primarily rodents. While these tests do not perfectly replicate the complexity of clinical pain conditions in humans, they provide valuable insights into the basic mechanisms of nociception (the perception of pain). They can serve as models for understanding certain aspects of pain processing. The hot plate and tail flick tests mimic acute pain and conditions involving temperature-induced pain. The tail flick test primarily assesses the spinal reflex response to thermal pain [38–40].
Cysteine, glycine, and glutamic acid compose the tripeptide GSH, which is present in most cells in remarkably high amounts. It plays a vital role in protecting cellular macromolecules against exogenous and endogenous reactive oxygen and nitrogen species [41]. Ferroptosis can be induced by molecules or situations that prevent the formation of GSH or GPX4, a GSH-dependent antioxidant enzyme [42]. It was discovered that morphine injection reduced the intracellular GSH level in the rat brain [43]. GPX4 is a specific type of selenoprotein with one selenocysteine residue at its active site and seven cysteine residues. GPX4 plays an important role in the regulation of ferroptosis, and inhibiting its activity promotes the occurrence of ferroptosis [29]. It is believed that a deficiency in GPX4 activity causes ferroptosis in neurodegenerative disorders [11]. Abdel-Zaher et al. [43] showed that prolonged injection of morphine into mice reduced the amount of intracellular GSH, a non-enzymatic antioxidant, and the activity of GPX, an enzymatic antioxidant. The Nrf2 is an increasingly recognized regulator that influences the vulnerability of cells to oxidative stress. Nrf2 plays a crucial role in maintaining the expression of various genes containing antioxidant response elements, both under normal conditions and when stimulated, to mitigate the detrimental effects of exposure to oxidants [44]. It has been established that Nrf2 is essential for controlling ferroptosis and for treating neurological diseases. Nrf2 can regulate the process of ferroptosis by controlling the levels of GPX4 protein, mitochondrial activity, and intracellular free iron [45].
It was shown that morphine treatment significantly influenced several antioxidant proteins involved in the Nrf2 pathway in HBMECs (human brain microvascular endothelial cells) [46].
Consistent with the above, the present findings demonstrate that repeated injections of morphine in rats led to the induction of oxidative stress and ferroptosis in the dorsal root ganglion tissues. This effect was confirmed by a decrease in GSH, GPX4, and Nrf2 levels. Otherwise, ferrostatin-1 suppressed the ferroptosis induced by repeated injection of morphine through increasing GSH, GPX4, and Nrf2 levels in the DRG. In line with the results of the present study, Chu et al. [17] demonstrated that ferrostatin-1 suppressed Glu-induced downregulation of GPX4 and Nrf2. In addition, An et al. [34] showed that ferrostatin-1 significantly enhanced GSH levels in dorsal root ganglion neurons injured by acrylamide. Ferrostatin-1 treatment significantly diminished the streptozotocin-induced decrease in GPX4 levels in the spinal cord [19]. Morphine exposure not only reduced the activities of antioxidants in target cells but also facilitated the production of free radicals, including ROS or reactive nitrogen species [47]. Avcı and Taşkıran [18] reported that morphine administration in single-dose and repeated doses decreased TAS levels and increased TOS levels in the DRG. This could indicate that morphine use inhibited the antioxidant system, potentially contributing to tolerance development. Consistent with the above, the present findings demonstrate that single and repeated injections of morphine in rats led to the development of oxidative stress in the dorsal root ganglion tissues. This effect was confirmed by a decrease in TAS levels and an increase in TOS levels. On the other hand, ferrostatin-1 suppressed the oxidative stress induced by morphine administration through increasing TAS levels and decreasing TOS levels in the DRG with morphine tolerance. In line with the present results, Chu et al. [17] showed that ferrostatin-1 can significantly reduce the levels of ROS in Glu-injured HT-22 cells. Additionally, Chen et al. [11] reported that liproxstatin-1, a ferroptosis inhibitor, markedly reduced the elevation of malondialdehyde and ROS levels induced by morphine in the serum and spinal cord of mice.
There are some shortcomings in this research, which are as follows:
The alteration in the cumulative antinociceptive effect of morphine by ferrostatin-1 was not assessed.
Despite findings from certain studies, such as those conducted by Zöllner et al. [48] and Blomqvist et al. [49], which suggested that chronic morphine administration might not always induce tolerance in the peripheral nervous system, the present study focused on investigating the specific changes occurring in the DRG. This particular area has been relatively less explored compared to central mechanisms, and the authors aimed to address this gap in the literature.
The paradigm used in this study involving naïve animals was indeed a limitation, and the authors acknowledge that it may not fully mimic the clinical condition where opioids are prescribed for pain relief in patients with pre-existing pain conditions or diseases. The authors aimed to lay the groundwork by investigating the effects of ferrostatin-1 on morphine tolerance under normal conditions, with a focus on the specific changes occurring in the DRG. It is also acknowledged that investigating the interaction of ferrostatin-1 with morphine in the context of disease conditions (e.g., nerve injury, complete Freund’s adjuvant-induced pain) would provide more meaningful insights into its potential clinical application. To advance the current understanding, it is essential to conduct further research using suitable pain models to investigate the interaction between ferrostatin-1 and morphine in the context of disease conditions.
Additional investigations are required to evaluate the effects of ferrostatin-1 on morphine's other side effects, including OIH (opioid-induced hyperalgesia).
We acknowledge the importance of investigating whether the effects of ferrostatin-1 are confined to the DRG or if they also extend to the CNS. Further studies exploring the involvement of the CNS and potential interactions with ferrostatin-1 are warranted.
Though thermal stimulus-based tests provide valuable insights into pain responses, the authors recognize that a combination of thermal and mechanical tests would have provided a more comprehensive evaluation of pain behaviors. The authors wish to encourage future studies, including their own, to incorporate both types of stimulus-based tests to further enhance the understanding of pain mechanisms and treatment modalities.
Conducting comprehensive research on various key pathways, such as apoptosis, nitric oxide pathway, and inflammation parameters, is essential to gain a better understanding of the drug's effects.
It has been noted that ferroptosis is mainly associated with iron deposition and involves key markers such as iron levels, ferritin, SLC7A11, and hepcidin. The authors acknowledge that the absence of these markers represents a limitation in the current study. Therefore, they are committed to addressing this limitation in future research endeavors.
Given the physiological distinctions between male and female rats [50], findings from male rats might not be directly applicable to females. Therefore, further research involving female animals is essential for comprehensive insights.
The markers investigated in this study may not provide a comprehensive understanding of the interplay between morphine tolerance and its modulation by ferrostatin-1 at the receptor level. Therefore, the authors plan to explore the effects of ferrostatin-1 on various aspects, including opioid receptor desensitization, internalization, oligomerization, and upregulation.
In conclusion, the results of this study suggest that ferrostatin-1 could reverse the development of morphine tolerance by suppressing ferroptosis and oxidative stress in DRG neurons, thereby proposing the potential therapeutic application of ferrostatin-1 to prevent or reduce the formation of morphine tolerance.
 XML Download
XML Download