

 PDF
PDF Citation
Citation Print
Print



Inflammatory idiopathic myopathies (IIMs) are uncommon but manageable conditions defined by muscle weakness and the presence of inflammatory cells, mainly T lymphocytes, in the muscle tissue. IIMs are categorized into five subgroups: dermatomyositis (DM), polymyositis (PM), inclusion body myositis (IBM), immune-mediated necrotizing myopathy (IMNM), and nonspecific myositis. These are all autoimmune disorders that are linked to distinct autoantibodies in different subcategories such as antisynthetase syndrome (ASS) and overlap myositis (OM). To conduct a comprehensive evaluation of myopathy patients, it is necessary to perform clinical assessment, electromyography, measurement of muscle enzymes, serological testing, imaging techniques, and histological muscle biopsies. Accurate subgroup classification is essential because of the diverse disease processes and therapeutic responses.
Certain subgroups exhibit positive responses to immunosuppressive medications. Immunohistochemistry is essential for diagnosing these disorders; however, there is ongoing discussion regarding the choice of a suitable panel for diagnosis. The objective of this study was to investigate the histological and immunohistochemical features of IIMs.
MATERIALS AND METHODS
The study involved a cohort of 56 Vietnamese individuals diagnosed with myositis at the Department of Pathology, University of Medicine and Pharmacy at Ho Chi Minh City. The research period spanned from January 1, 2019, to June 30, 2023. Sample selection criteria were cases diagnosed with myositis based on clinical assessments that adhered to the categorization criteria specified by the European League Against Rheumatism (EULAR) in 2017, with a probability surpassing 55% (particularly, those classified as definite or probable).
We categorized patients into one of six subgroups based on their ultimate clinical presentation, antibody panel results obtained through immunoblot assay, and findings from muscle biopsy. Furthermore, we adhered to the diagnostic criteria outlined for DM by the European Neuromuscular Centre (ENMC) in 2018 [1], for IMNM by the ENMC in 2016 [2], and for IBM by the ENMC in 2011 [3].
Exclusion criteria encompassed evidence indicating other causes of myopathy such as drug-induced myopathy, exposure to toxic substances, infectious myopathy, endocrine disorders, or severe neurological disorders in internal medicine conditions. Additionally, evidence from family history, clinical characteristics, genetic testing, or histopathology suggestive of genetic etiology was considered.
Muscle biopsy samples were preserved using liquid nitrogen and isopentane, followed by staining with hematoxylin and eosin, modified Gomori Trichrome, periodic-acid Shiff, and NADH. Immunohistochemistry was conducted on frozen sections using the following panel of antibodies: HLA-ABC (W6/32, Invitrogen, Carlsbad, CA, USA), HLA-DR (LN3, Invitrogen), C5b-9 (aE11, Invitrogen), Anti-SQSTM1/p62 (GT1478, Invitrogen), and Mx1/2/3 (sc166412, Santa Cruz, Dallas, TX, USA).
The study collected clinical data of age, sex, sites of muscle weakness, muscle strength, creatine phosphokinase (CK) level in the blood, electromyography findings, and specific autoantibodies recorded in the pathology requisition form. Histological and immunohistochemical analysis was used to evaluate the presence and distribution of inflammatory cells in muscle tissue, as well as the occurrence of perifascicular atrophy (PFA), necrosis, phagocytosis, and rimmed vacuoles.
The Pearson’s χ2 test (or Fisher’s exact test) was used to evaluate the relationship between pairs of categorical variables. All statistical analyses were performed using SPSS ver. 20.0 (IBM Corp., Armonk, NY, USA). A p-value <.05 was considered statistically significant.
RESULTS
Clinical characteristics of inflammatory myopathies
According to our records, there are six categories of inflammatory myopathy with the following numbers of cases and percentages as determined in this study: IMNM 33 (58.9%), DM 13 (23.2%), OM 5 (8.9%), ASS 3 (5.4%), IBM 1 (1.8%), and PM 1 (1.8%). The average age was 49.7–16.1 years, with the youngest patient being 17 years old and the oldest being 79. The age with the highest disease prevalence was 43 years. The majority of patients was female, accounting for 73.2% of the cases. The female-to-male ratio was 3:1.
The average duration from symptom onset to diagnosis was six months, with no notable distinction between groups, except for cases of IBM, which exhibited an extended diagnostic period of 36 months. While IBM is characterized by a prominent manifestation of distal weakness in the upper limbs, the other groups predominantly displayed proximal weakness.
Skin lesions, a hallmark of DM and also present in ASS, were documented in 67% of cases. In the DM subgroup, 100% of cases presented with skin lesions, among which specific lesions (heliotrope sign, Gottron’s sign, and Gottron’s papules) were observed in seven of 13 cases (53.8%). The remaining skin lesions included poikiloderma (38.4%), V-sign (46.1%), mechanics’ hands (46.1%), and nonspecific lesions (46.1%). In the ASS subgroup, all three patients (100%) had mechanic’s hands, and one case also exhibited Gottron’s sign, while two cases had nonspecific skin lesions (Table 1).
Difficulty in swallowing was noted in 16% of cases. Pulmonary involvement, as diagnosed by computed tomography scan, was observed in 21% of cases, with respiratory failure more commonly occurring in the DM, ASS, and OM groups compared to the IMNM group. The mean blood CK level at the time of diagnosis was 3,997 U/L. The IMNM group exhibited a higher CK level in the blood compared to the DM group.
In serological assessments, the diagnostic positivity rate was 32 of 43 cases (74.4%). Among them, nine cases were positive for more than two antibodies (excluding Ro-52), exhibiting distinct clinical features associated with MDA5-PL7, Mi2-SRP, NXP2-SRP, PM-Scl-EJ, and PM-Scl-Jo1. Additionally, 23 cases (53.5%) positive for a single antibody contributed to the classification of myositis subgroups.
Pathological findings of inflammatory myopathies
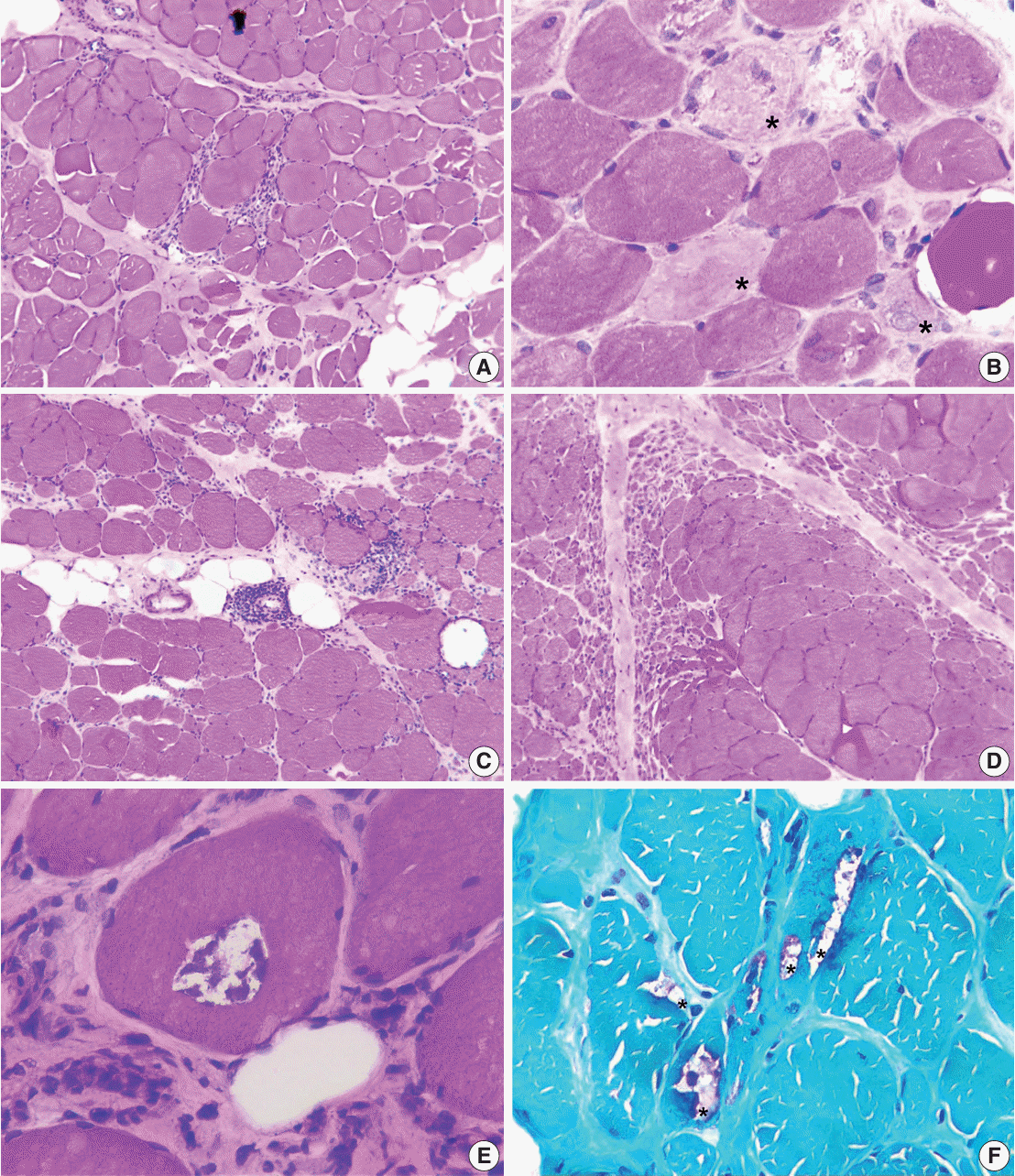
Among the total 56 cases, the described pathological features on biopsy had diagnostic and classificatory significance for inflammatory myositis. PFA was found in 10 cases (17.8%), in 100% of the ASS group and 50% of the DM group. Infiltration of lymphocytes was observed in 62.5% of the cases, with the highest frequency in the OM group with five cases (100%) and the lowest in the IMNM group. Fiber necrosis was observed in 75.0%, with the highest percentage in the ASS group (100%), 93.9% in the IMNM group, 23.1% in the DM group, 80.0% in the OM group, and zero in the IBM group. Endomysial fibrosis was noted in 23% of the cases. Vasculitis was detected in 5.3% of the cases, with the highest incidence in the OM group (40.0%). Rimmed vacuoles, a distinctive feature of IBM, were identified in only one case of IBM in our study (Table 2, Fig. 1).
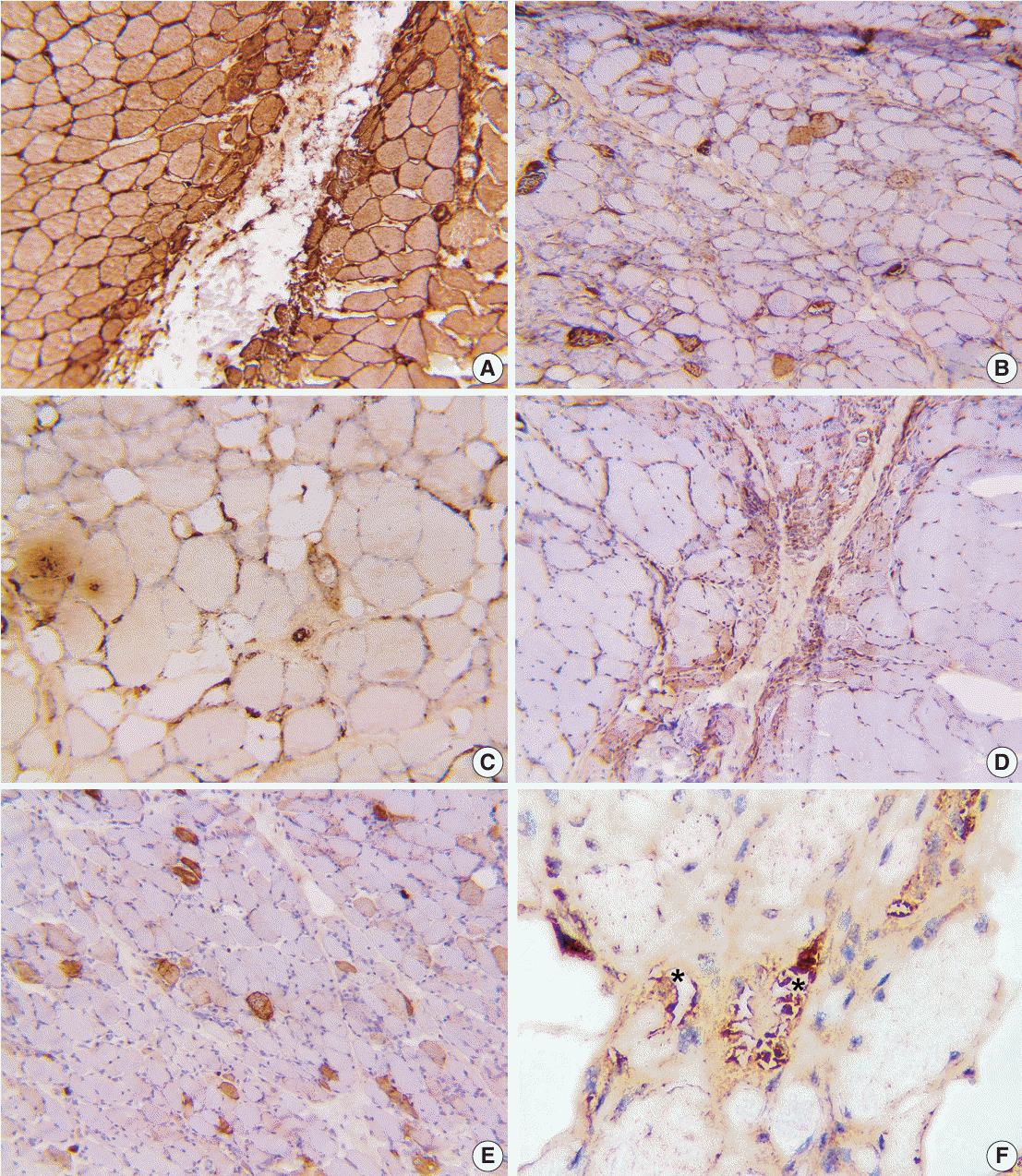
Regarding immunohistochemical staining, major histocompatibility complex I (MHC-I, HLA-ABC) expression was noted in 89% of the cases. Abnormal expression of MHC-II (HLA-DR) was observed in 19.6% of the cases. Mx1/2/3 (MxA), a distinctive marker for DM, showed abnormalities in 10.7% of the total cases, all of which belonged to the DM and ASS groups. In 57% of the cases, we observed abnormal membrane attack complex (MAC) expression in the sarcolemma and endomysial capillaries, with C5b-9 deposited on capillaries and perivascular inflammation noted in 25% of cases. Additionally, MAC expression was observed in muscle fiber necrosis, serving as a nonspecific marker. However, there was no significant difference in MAC deposits between PM and DM, while p62 expression was noted in the one IBM case (Table 3, Fig. 2).
The combined use of MHC-I and MAC can identify 96% of inflammatory myositis cases. Among the cases examined, only two DM cases (3.5%) exhibited no expression of MHC-I, MHC-II, MAC, Mx1/2/3, or p62.
DISCUSSION
Regarding the disease subgrouping in our study, the highest proportion of patients were in the IMNM group (58.9%), which is consistent with the study conducted by Watanabe et al. [4]. In the Watanabe study, the DM subgroup had a higher prevalence compared to our study. Conversely, the OM subgroup had a higher prevalence in our study compared to the Gupta et al. [5] and Ohnmar et al. [6]’s studies. Only one case was categorized into the IBM subgroup in our study, and none were reported in the studies by Gupta et al. [5] and Ohnmar et al. [6]. In contrast, IBM cases accounted for 16% of the Watanabe et al.’s study [4]. This difference could be due to the smaller sample size in our study as well as variations in ethnic characteristics. IBM is a more common subgroup in the white population over 50 years of age. Clinical features of the IBM subgroup include distinct weakness patterns involving upper and lower limb muscles and a slowly progressive course over many years, making it challenging to diagnose, with common misdiagnosis.
The age of onset in our study exhibited a standard distribution, with a mean age of 50 and a peak disease frequency at 43 years of age, which is consistent with the study by Chen et al. [7], who conducted a retrospective population-based study on the Chinese population with a mean age of 51.2 years. The mean age in our study was slightly higher compared to the study by van der Meulen et al. [8]. Among the inflammatory myopathy subgroups, age of onset did not show significant differences. This finding is in line with other epidemiological studies [5,8-10].
The infiltration of lymphocytes into the biopsy tissue is a characteristic feature initially described in the histopathological criteria for DM diagnosis by Bohan and Peter [9] in the 1970s. However, this phenomenon can also be encountered in other conditions characterized by muscle fiber breakdown, such as Duchenne or Becker muscular dystrophy. Lymphocytic infiltration may not be detected in cases of amyopathic DM, and it can be absent in patients who have received prior immunosuppressive therapy. In cases with nonspecific findings on muscle biopsy, the diagnosis of OM in our study primarily relied on autoantibody testing (positive for PM-Scl and anti-Ku) and clinical features indicative of multisystem involvement, muscle stiffness on examination, and evidence of systemic vasculitis.
PFA, which is specific for DM, was observed in 46% of the DM cases in our study, which is lower than the study conducted by Uruha et al. [10]. Consensus on the diagnostic criteria for DM from the ENMC indicates that approximately 50% of cases exhibit this feature [1]. The lower PFA rate observed in our study could be linked to expertise and biopsy site selection. Utilizing Mx1/2/3 immunohistochemistry is instrumental in improving the identification of PFA fibers, which highlights the crucial role of Mx1/2/3 immunohistochemistry in the diagnosis of DM.
PFA is specific for DM; however, it also can be observed in ASS. According to the findings of Uruha et al. [10], PFA was observed in 13% of ASS cases. Mx1/2/3 or MxA expression helps differentiate between DM and ASS when both are present with PFA. In our study, no cases of ASS tested positive for Mx1/2/3. These results align with the study by Inoue et al. [11], who observed similar clinical manifestations between DM and some ASS cases but found no positivity for Mx1/2/3.
In our study, fiber necrosis was observed in 42 of 56 cases (75%). The prevalence of fiber necrosis varied among subgroups as follows: IMNM (93.9%), DM (23.1%), OM (80.0%), ASS (100%), and PM (100%) and was not observed in the IBM subgroup. The degree of fiber necrosis was also notably higher in the IMNM group compared to the DM group (94% vs. 23%). In the ASS subgroup, fiber necrosis was consistently observed at a very high rate (100%), surpassing the 48% reported in the study by Noguchi et al. [12].
Based on immunohistochemistry, our study recorded the following positivity rates: HLA-ABC (MHC-I, 89.2%), HLA-DR (MHC-II, 19.6%), C5b-9 (MAC, 57.1%), and Mx1/2/3 (10.7%), notably all of positive Mx1/2/3 staining cases were DM. MAC deposits were identified in endomysial capillaries in 25% of cases. Our study showed that MHC-I expression had the highest sensitivity for detecting abnormalities, while MHC-II, Mx1/2/3, and MAC showed lower positivity rates but higher specificity in myositis sub-classification. Compared with previous studies by Das et al. [13], Rider et al. [14], and Uruha et al. [15], our study showed similarity in the positivity rate of MHC-I, while the other markers exhibited differences mainly due to variations in the initial classification criteria. The Das et al.’s [13] and Rider et al.’s [14] studies only classified PM and DM in their diagnosis, and Uruha et al.’s study [15] primarily relied on antibody screening for the initial sample selection.
While MHC-I has been demonstrated to have high sensitivity, this immunohistochemical staining method can yield unusual results in various myopathies due to different underlying causes. According to van der Pas et al. [16], 11% of cases with dysferlinopathy tested positive for MHC-I, as did 4% of other non-inflammatory myopathies. Another study by Confalonieri et al. [17] found relatively high rates (70%) of dysferlinopathy with MHC-I positivity and 20% with MHC-II positivity, but no cases of Duchenne muscular dystrophy tested positive. Many other studies have reported varying positivity rates, demonstrating that, while MHC-I has high sensitivity, it may not be highly specific. In a retrospective study by Rodriguez Cruz et al. [18] analyzing biopsy samples from groups with and without inflammation (inflammatory myopathies, non-inflammatory myopathies, genetic myopathies, drug-induced myopathies, severe medical conditions), they observed 98% positivity for MHC-I in inflammatory myopathies and 92% positivity in non-inflammatory myopathies. For MHC-II, they found 60% positivity in inflammatory myopathies and 10.1% in non-inflammatory myopathies. These rates, in comparison to our study, show similarity in MHC-I positivity and lower MHC-II positivity. Notably, no cases in that previous study were MHC-II positive without MHC-I positivity, which aligns with our findings.
The analysis of inflammatory cells, MHC-I expression, and MAC deposits plays a role in distinguishing dysferlinopathy from IIMs [19]. In our study, the combined use of MAC and MHC-1 was effective in identifying 96% of inflammatory myositis cases in muscle biopsies. Another study indicated that MAC deposits on capillaries were observed in childhood DM, suggesting that MAC deposits on endomysial capillaries could serve as a valuable indicator of early-stage DM [20].
Of the cases studied, only two DM cases (3.5%) did not exhibit positivity with any immunohistochemical staining method. However, the presence of other clinical features and biochemical abnormalities also contributed to the final diagnosis in these cases. This underscores the importance of a combination of clinical parameters and biopsy for an accurate diagnosis. It is essential to consider the location of muscle biopsy as cases with extensive fibrosis may not be helpful for diagnosis.
The sample size for the current study is limited, and several subgroups have a small number of cases, possibly not fully representing the groups in terms of clinical and laboratory characteristics, especially for the Vietnamese population. Nevertheless, considering the rarity of these diseases, the initial assessments based on pathological and immunohistochemical findings offer neurologists valuable insights for diagnosis and treatment. Still, immunohistochemical staining supports the diagnosis, and muscle biopsy aids in diagnosing specific muscle diseases, accurately classifying the group of inflammatory myopathies in close alignment with clinical findings.
The integration of muscle biopsy and antibody testing is essential for accurately categorizing and diagnosing myositis, as it is a highly sensitive clinical procedure. To diagnose inflammatory myopathies through muscle biopsy, it is essential to perform immunohistochemistry with a variety of markers, including MHC-I, MHC-II, C5b9, and Mx1/2/3. p62 staining is essential in cases of suspicion of IBM. The diagnosis of myositis is, however, a multi-modal process, where pathology and immunohistochemical staining play a supporting role. Successful pathological diagnosis of myositis may vary depending on the biopsy location, the condition of the biopsy sample, and the expertise of the pathologist.
 XML Download
XML Download