

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Cardiovascular diseases (CVDs) are a preeminent global cause of morbidity and mortality. Different types of cells orchestrate intricate roles in processes integral to the pathophysiological panorama of CVDs, such as cardiac dysfunction, myocardial fibrosis, and ventricular remodeling, during its development and progression [1,2]. Early detection, preventative strategies, and appropriate medical interventions are essential in diminishing the burden of CVDs and supporting the integrity of the circulatory milieu. Metabolism refers to an intricate sequence of biochemical reactions within the organism to convert nutrients into energy for growth, repair and various physiological processes. However, certain metabolic disturbances interfere metabolic homeostasis, leading to abnormal transformation and decomposition of amino acids, carbohydrates, or lipids, and may also impact the mitochondrial proficiency in energy production, culminating in multiple metabolic disorders, such as diabetes, obesity, and dyslipidemia [3-5]. Gaining insights into the underlying mechanisms that drive the progression of CVDs and metabolic diseases, particularly under the context of distinct cell types, assumes paramount importance for elucidating the pathophysiological intricacies as well as developing effective treatment options [2].
Protein arginine methyltransferases (PRMTs), a class of enzymes responsible for the precise regulation of protein methylation, perform critical roles in the pathophysiology of CVDs and metabolic diseases [6,7]. In-depth investigation of PRMTs functional value is imperative for identifying the molecular mechanisms, contributing to unveiling novel insights into disease progression, while concurrently paving the way to innovative therapeutic interventions. In this context, this review aims to intricately delineate the multifaceted contributions of PRMTs to both the cardiovascular system and metabolic ailments, placing particular emphasis on the nuanced functions exhibited across diverse cellular contexts. We endeavor to provide a comprehensive understanding of the involvement of PRMTs in the etiology of diseases and their potential as promising therapeutic targets.
ARGININE METHYLATION AND PRMTs
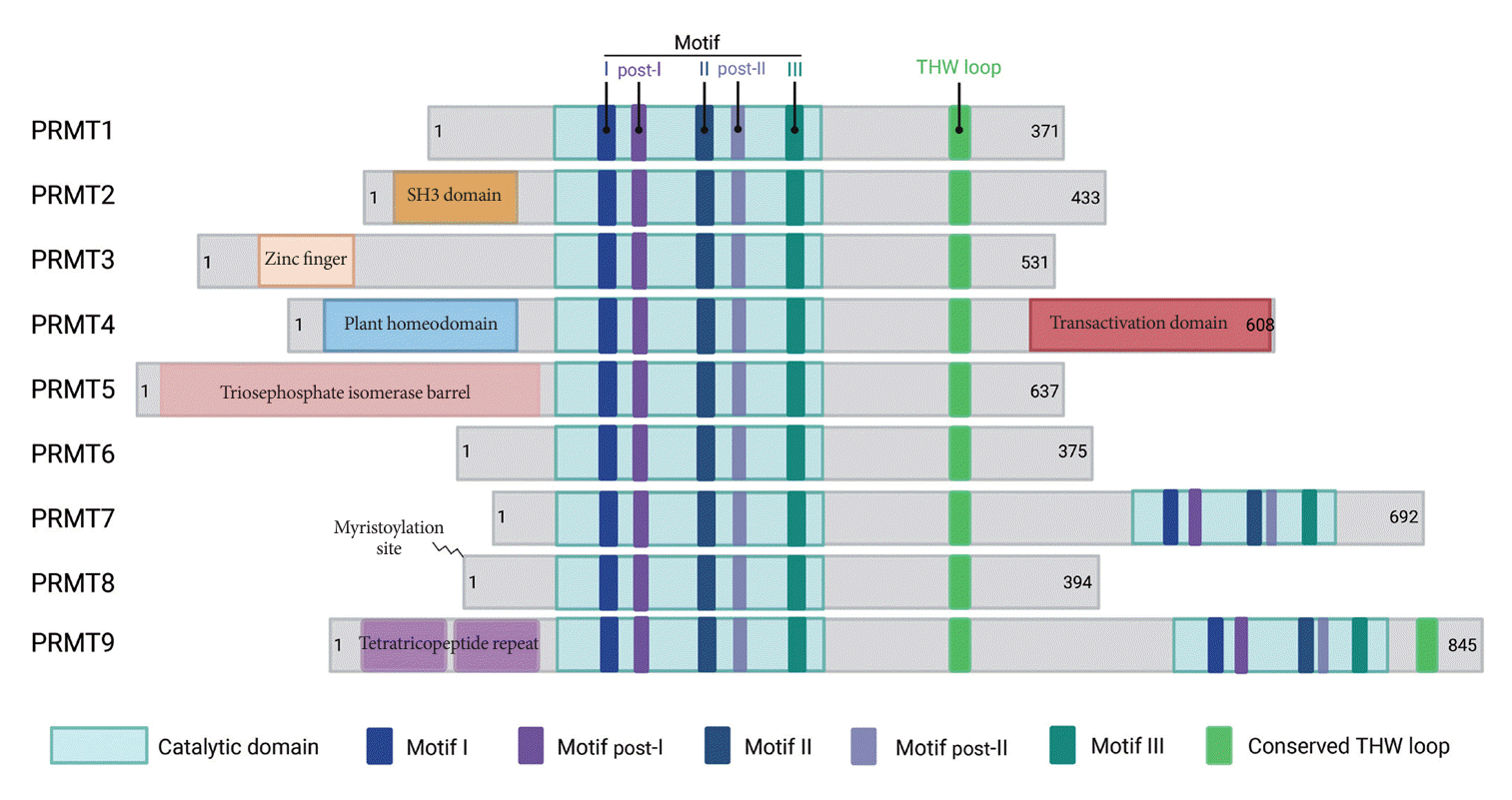
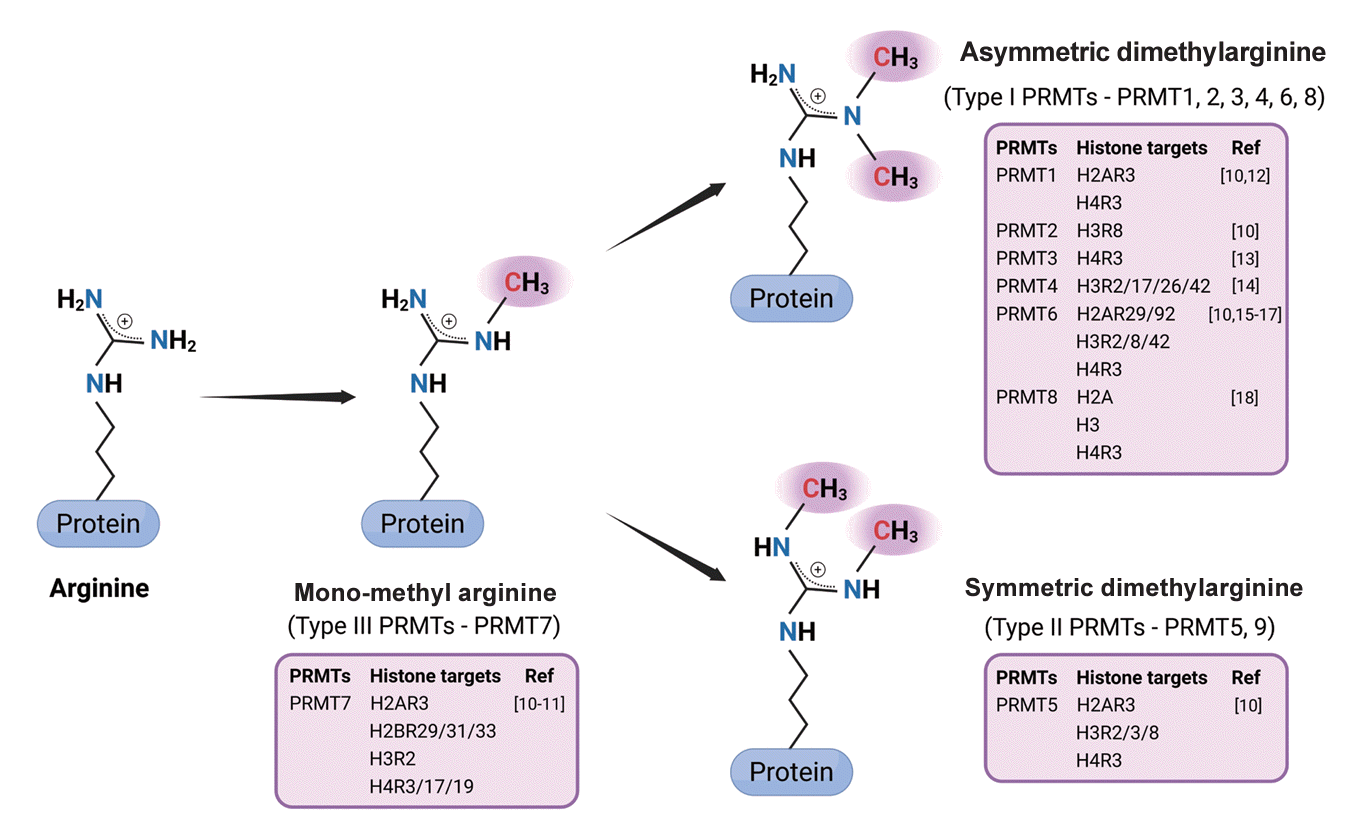
Arginine methylation stands out as the distinctive post-translational modification involving the addition of a methyl group to arginine residues. The orchestration of arginine methylation is chiefly executed by PRMTs, which function as molecular hubs in this context. In mammals, PRMTs constitute a family of nine annotated members, all of which share conserved catalytic domains with a high sequence similarity (Fig. 1). Based on their catalytic activity, PRMTs are categorized into three types. Both type I and type II PRMTs utilize monomethylarginine (MMA) as an intermediate, proceeding with subsequent catalytic processes to form symmetric or asymmetric methylated arginine. Type III PRMT, exemplified by PRMT7, exclusively produces MMA. Type I PRMTs, which include PRMT1/2/3/4/6/8, catalyze the transfer of two methyl groups to the nitrogen atom of arginine with an asymmetric manner which form the asymmetric dimethylarginine (ADMA). PRMT5/9 are type II PRMTs which catalyze the transfer of two methyl groups onto the arginine residue and undergo dimethylation, resulting in the symmetric addition of two methyl groups on both sides (Fig. 2) [8]. Notably, PRMTs exhibit diverse cellular localization, with some (PRMT6/9) predominantly enriched in the nucleus, orchestrating critical roles in gene expression and chromatin regulation. Others (PRMT3) reside primarily in the cytoplasm, influencing protein translation and signaling pathways regulating cell behavior. Certain PRMTs (PRMT1/2/4/5/7) dynamically shuttle between both compartments, showcasing versatile functions in various cellular contexts. While PRMT8 is mainly expressed in the plasma membrane within the neuronal system, where it plays significant roles in brain development [9]. PRMTs wields a profound impact, not only on gene expression by histone modulation (Fig. 2) [10-18], but also on various non-histone substrates that participate in essential biological phenomena [19], encompassing cell signaling, transcription and translation processes, DNA damage response, pre-mRNA splicing, and protein stability control [7].
Latest research has underscored the predominance of PRMTs and their aberrant modulation in the pathogenesis of various human diseases, including cancer, CVDs, metabolic disruptions, neurological disorders, and viral infections [19,20]. In this case, their therapeutic potentials are being investigated in preclinical and clinical settings. However, the complicated biological features of PRMTs and their downstream substrates, on the contrary, indicate challenges and potential restrictions in the realm of targeted therapeutics, which are under ongoing but limited unveiling. The indispensable roles of PRMT1/5 in embryonic development have been revealed by research using knockout (KO) mouse model [21-25]. In the case of other PRMT subtypes, deficiencies show a spectrum of symptoms, ranging from neonatal death to developmental delays [26-28]. Tissue-specific KO models further benefit us in understanding the physiological activities of each PRMT isoform. However, due to presence of functional redundancy and substrate specificity, determining the precise effects of individual PRMTs remains challenging. Further research is imperative to fully unravel the activities of each PRMT subtype and to explore the therapeutic potential of arginine methylation in the context of various disease treatments.
PRMTs IN CARDIOVASCULAR DISEASES
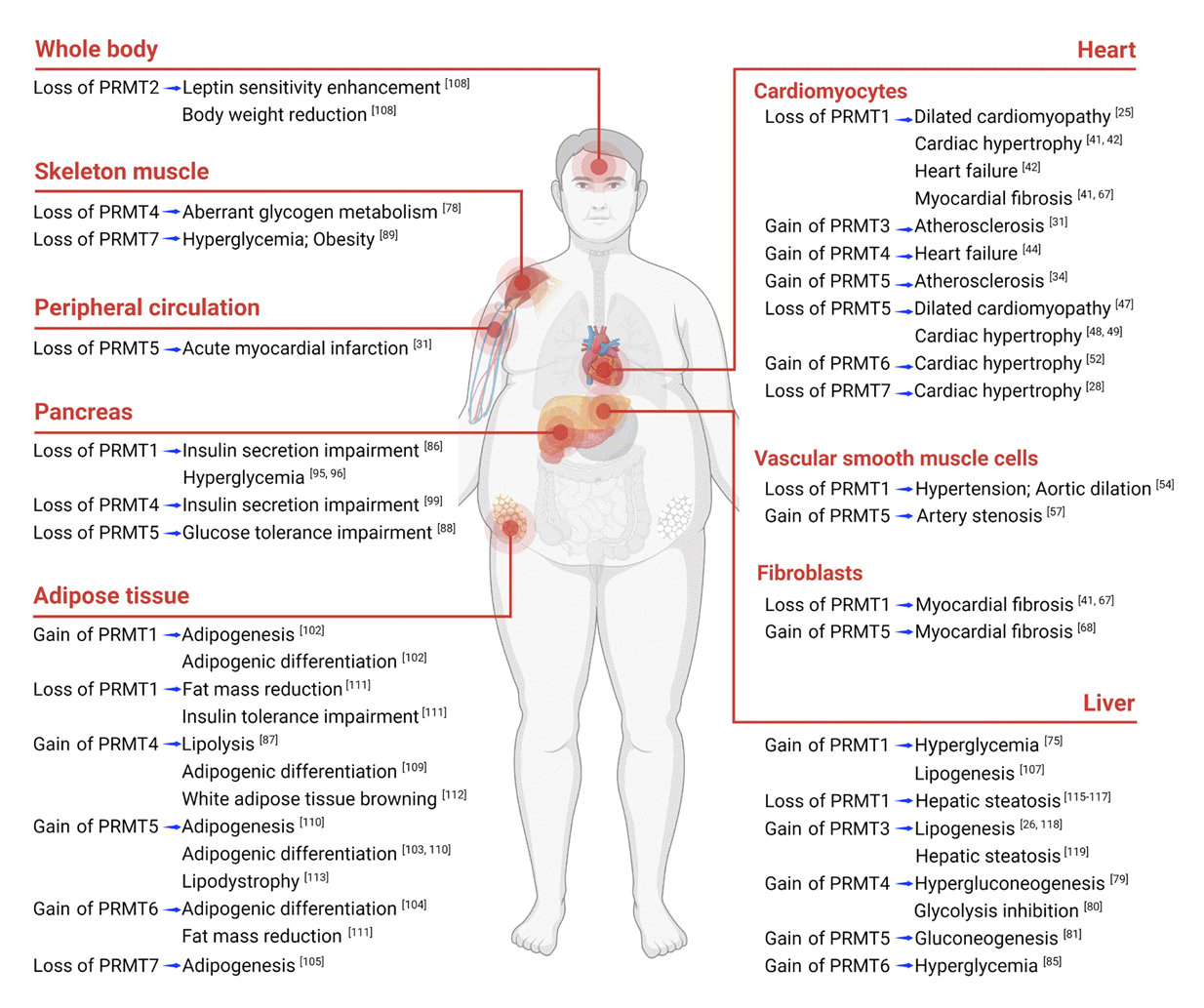
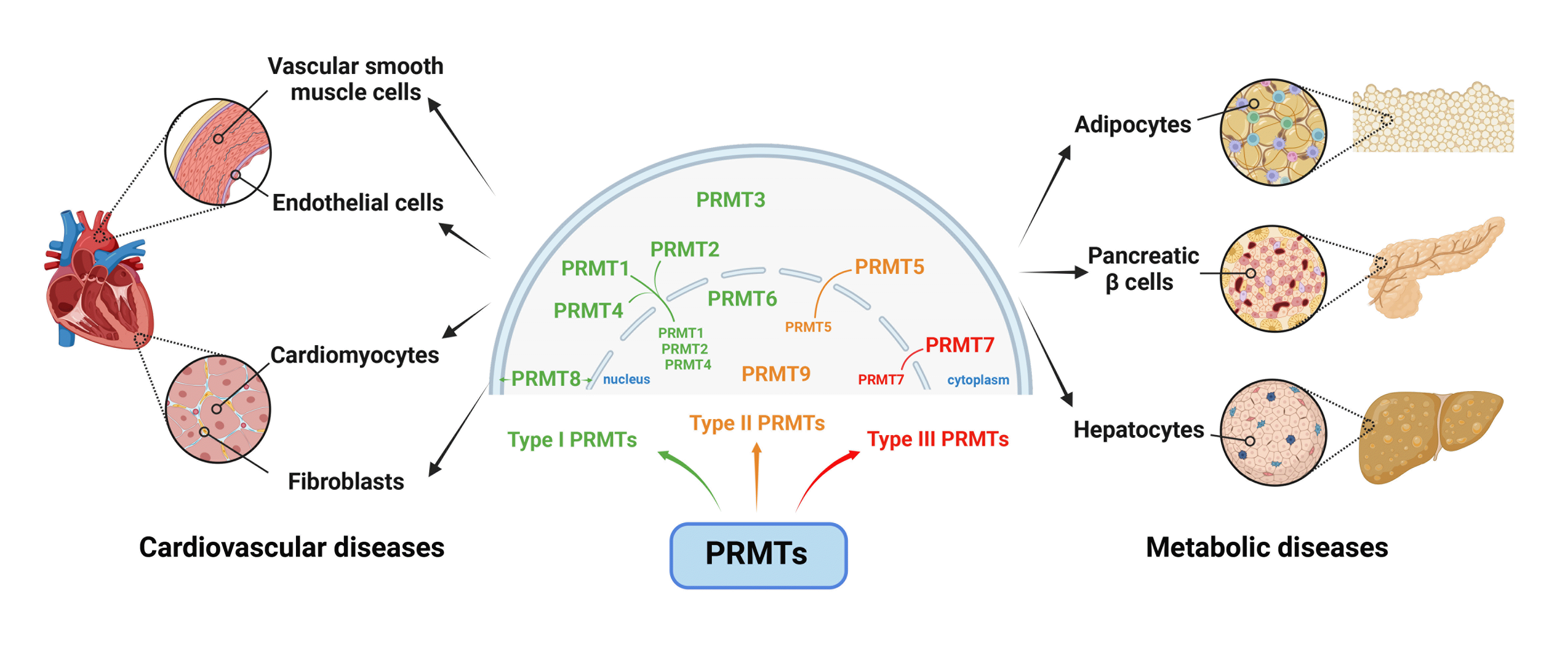
PRMTs have been widely proved as pivotal contributors in both physiological and pathological regulation of CVDs (Fig. 3). For instance, PRMTs dysregulation precipitates endothelial dysfunction, resulting in increased permeability, inflammatory response, and aberrant vasodilation, thereby culminating in atherosclerosis [29-32]. Besides, PRMTs actively participate in blood pressure regulation by influencing vascular tone and endothelial function [33,34]. PRMTs also affect proteins responsible for vascular smooth muscle contraction and relaxation, thereby determining blood vessel diameter and blood pressure regulation [35,36]. Additionally, PRMTs affect proteins integral to endothelial cell (EC) signaling, nitric oxide (NO) production, and vascular reactivity, hence impacting both endothelial health and dysfunction [37-39]. Moreover, PRMTs are revealed as pivotal regulators governing cardiac repolarization through modulating interactions among ion channel [40]. Under these circumstances, it is imperative to recognize that the effects of PRMTs in the realm of CVDs are characterized by their complexity and specificity under diverse conditions. Unmasking the specific mechanisms of PRMTs in each cell type assumes paramount significance, holding the promise of clarifying targeted therapeutic interventions in effectively combating CVDs.
Cardiomyocytes
Cardiomyocytes, the primary constituents of cardiac muscle tissue, are responsible for the contraction and pumping function of the heart. Current research has provided compelling evidence linking PRMT1 within cardiomyocytes to the pathogenesis of heart failure (HF). PRMT1 expression was downregulated in individuals afflicted with HF [41]. PRMT1 deficiency evoked hypertrophic responses and remodeling gene expression, whereas elevated PRMT1 suppressed pathogenic reactions to neurohormones in isolated cardiomyocytes, suggesting PRMT1 performed protective effects in HF progression. Certain assumptions were substantiated by multiple mouse models featuring cardiomyocyte specific PRMT1 ablation, which had exhibited typical characteristics of HF such as ventricular enlargement and declined contractile function [25]. This absence also affected mRNA alternative splicing in the heart, highlighting the significance of PRMT1 in regulating alternative splicing processes and maintaining homeostasis. In our previous investigation, we demonstrated a rapid progression to dilated cardiomyopathy (DCM) within a mere two-month timeframe in mice lacking PRMT1 specifically within cardiomyocytes, accompanied by cardiomyocyte hypertrophy, cytoskeletal disruption, and fibrotic alterations [41]. The underlying mechanism was related with the Ca2+/calmodulin-dependent protein kinase II activity regulated by PRMT1 via methylation at Arg 9 and 275, which emphasizing the significance of arginine methylation as a pivotal mechanism under the content of HF and cardiomyopathy.
PRMT1 in cardiomyocytes also closely relates with endoplasmic reticulum (ER) stress response and cell death, in terms of both disease progression and drug induction. Our prior studies revealed that hearts with PRMT1 deficiency demonstrated aberrant cell death in early life stage. Triggering transcription factor 4 (ATF4)/C/EBP homologous protein (CHOP) pathway, a pivotal regulator in the unfolded protein response activated in response to ER stress, have been proved to be activated in neonatal rat ventricular myocytes and cardiomyocytes when PRMT1 was knockdown or interfered [42]. PRMT1-deficient hearts expressed similar overall gene expression profiles as the wild-type hearts subjected to the ER stress inducer tunicamycin, while PRMT1-deficient heart showed exacerbated cell death after tunicamycin therapy. Along with these findings, we also discovered that PRMT1 against ER stress was also seen in hearts with doxorubicin (Dox)-induced cardiotoxicity [42]. The inhibition of PRMT1 gene expression or enzymatic activity exacerbated Dox-induced cardiomyocytes injury. Conversely, PRMT1 overexpression demonstrated a decline in the extent of cell death and reactive oxygen species (ROS) production [43]. Transient exposure of cardiomyocytes to Dox prompts an upsurge in PRMT1 activity and its relocation to the ER. Notably, enhanced PRMT1 expression has been shown to suppress the ER stress response by facilitating the methylation of activating ATF4, suggesting a plausible mechanistic basis for PRMT1’s protective attributes.
PRMT4 is essential for modulating cardiomyocyte apoptosis in the pathological process of cardiac ischemia such as myocardial infarction (MI). In ischemia heart and hypoxic cardiomyocytes, PRMT4 significantly overexpressed. In line with this, cardiac-specific PRMT4 overexpression deteriorated left ventricular function and worsened cardiac remodeling postMI, primarily attributed to aggravated cardiomyocytes apoptosis [44]. However, on the contrary, PRMT4 expression is significantly downregulated in hearts exposed to Dox [45]. It’s worthy notion that overexpressed PRMT4 aggravated ferroptosis to exacerbate Dox-induced cardiotoxicity, whereas its pharmaceutical inhibition or gene disruption displayed the adverse impact. The underlying mechanism involved PRMT4 inhibiting nuclear factor erythroid 2-related factor 2/glutathione peroxidase 4 (Nrf2/GPX4) signaling to exacerbate ferroptosis and subsequent Dox-induced cardiomyopathy. These contradictory findings underscore the intricate regulatory implication of PRMT4 in cardiomyocytes, which may necessitate further exploration to clarify the underlying mechanisms in-depth, and to elucidate the possible connections between diverse regulations.
Most arginine residues in cells undergo symmetrical dimethylation through the action of PRMT5, which also has a variety of functions in signal transduction, protein function, and transcriptional regulation [46]. Despite the significance of PRMT5, its precise functional roles and mechanisms in the heart are relatively poor understood. Recent research has brought to light the significance of PRMT5 in cardiac biology, particularly in the context of DCM. Mechanistically, PRMT5 regulated protein O-GlcNAcylation, a key regulator of DCM modulated by O-GlcNAcase (OGA), to maintain cardiac homeostasis. PRMT5 deficiency inhibited OGA expression by triggering aberrant splicing, thereby culminating to DCM, which was reversed by cardiac OGA overexpression [47]. In addition to DCM, PRMT5 has been found to perform protective effects on CH, with a declined expression in either in vitro or in vivo hypotrophy models. PRMT5 functioned as a crucial epigenetic regulatory factor by enhancing H4R3me2s, with its silencing or activity inhibition enhanced gene expression related to CH, while its overexpression reversed the effects [48]. Consistent finding was evidenced from the research of PRMT5 on NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome in rat hypertrophy model, where PRMT5 was revealed to ameliorate angiotensin II (Ang II)-stimulated cardiac hypertrophy (CH) through E2F-1/nuclear factor κB (NF-κB) signaling pathway [49]. This finding emphasized the significant regulatory effects of PRMT5 in this pathological alteration, while implying the potential values of PRMT5 in inflammatory apoptosis and pyroptosis progression, which warrant further exploration. In line, elevated PRMT5 mechanically inhibited the acetylation of GATA binding protein 4 (GATA4), a pivotal contributor of CH, and attenuated hypertrophic responses in phenylephrine (PE)-induced cardiomyocytes, whereas PRMT5 knockdown led to the activation of GATA4 and consequent cardiomyocyte hypertrophy [50]. Apart from these findings, PRMT5 has also been implicated in oxygen and glucose deprivation renal reperfusion injury via methylation mediation related with pyroptosis [51], whose regulatory effects in ischemic heart remain unclear and require further evaluation. Collectively, PRMT5 is expected to be developed as a promising therapeutic target against multiple cardiac pathological alterations.
Myocardium seldomly expressed PRMT6, leading to a lack of wild studies. However, current available research has displayed significant impacts of PRMT6 on the heart. Consistent with PRMT5, PRMT6 was also revealed to perform regulatory effects on CH but are opposite in nature. PRMT6 exhibited a consistent increased expression pattern in CH induced by various inducements, such as in surgery- and PE-induced animal models, as well as patients with end-stage HF [52]. Mechanical study evidenced PRMT6’s methylation on histone H3 arginine 2 asymmetric demethylation (H3R2Me2a) and revealed its potential roles in modulating cardiomyocyte contraction and electrophysiology. Further research is imperative to explore additional implications of PRMT6 in other CVDs.
In according with PRMT5, PRMT7 also functioned as a protective factor in CH pathological progression. Our prior studies proved the deficiency in PRMT7 contributed to CH and fibrosis, and worsened Ang II-induced CH [28]. Conversely, PRMT7 overexpression reversed this phenotype, through the mechanisms of methylating Arg 93 on the β-catenin protein to inhibit the Wnt signaling pathway activity. Meanwhile, our ongoing projects display a worsen cardiac dysfunction in mice with PRMT7 ablation post-MI modeling, suggesting the potential effects of PRMT7 in maintaining cardiac homeostasis and promoting cardiac recovery.
Vascular smooth muscle cells
Vascular smooth muscle cells (VSMCs), primarily located in the middle layer of blood arteries, regulate vascular contraction [53]. Targeted ablation of the PRMT1 gene triggered VSMCs death and the deterioration of elastic fibers, culminating in various disorders, such as hypotension, aortic dilation, and aortic dissection [54], This cascade suggests a systematic dysfunction affecting vascular structure, elasticity, and stability. Notably, the aortas of elder individuals and those with aortic aneurysms exhibited a considerable downregulation of PRMT1 as well as genes associated to contractility. PRMT1 ablation, in turn, increased the synthetic-related genes expression while decreased the contractility-related genes expression such as myocardin, which contributed VSMCs to shift from contractile to synthetic phenotype, leading to the functional decline in aortic contractility and VSMC traction force [54]. Ang II significantly reduced PRMT2 expression in VSMCs at both transcription and translation levels, whereas PRMT2 overexpression inhibited Ang II-induced VSMCs proliferation and inflammation [55], indicating the protective implication of PRMT2 in VSMCs homeostasis. In contrast, PRMT5 has been found to positively link to vascular inflammation, with PRMT5 stimulated the production of vascular cell adhesion molecule 1 (VCAM1) by demethylating Arg 30 on the NF-κB protein. In according, PRMT5 specific deletion in VSMCs diminished vascular inflammation and decreased VCAM1 expression levels [56]. Moreover, PRMT5 also positively associated with vascular contraction decline and neointimal hyperplasia, evidenced by its aberrant overexpression in clinical carotid artery stenosis. PRMT5 deletion in VSMCs exhibited elevated contractile markers, while, on the contrary, excessive PRMT5 expression encouraged opposite outcomes [57]. PRMT5 has also been indicated in vascular remodeling, in the mechanism of promoting VSMCs phenotypic switching through the inhibition of ubiquitin-dependent proteolysis of Kruppel-like factor 4. PRMTs emerge as critical regulators in maintaining VSMC structure and function, and are involved in distinct cardiac pathogenesis, providing promising therapeutic possibilities. However, relevant research in this domain is largely limited, highlighting a great demand of further mechanistic studies.
Endothelial cells
ECs, located in the inner layer of blood vessels, are vital for regulating vascular relaxation, sustaining blood flow, and producing bioactive substances like NO [58,59], functioning crucial modulation in cardiac physiological states. Lipopolysaccharide exposure is well-established to induce ECs activation via NF-κB and mitogen-activated protein kinases. Recent studies unveiled the implications of PRMT1 in this process, with an upregulated mRNA expression as well as intracellular ADMA concentrations, promoting cellular injury and vascular inflammation, indicating the attributes of PRMT1 in endothelial dysfunction [60]. Furthermore, prior investigations have highlighted the role of PRMT1 in triggering ECs dysfunction observed in the context of high salt. The involved mechanisms are intricately linked to PRMT1-induced elevation in ADMA expression, which subsequently induced suppression of endothelial NO synthase biosynthesis [61]. These findings, in conjunction with aforementioned observations, unequivocally affirm the multifaceted complex role of PRMT1 in CVDs, which underscores the imperative necessity to delve into the effects of PRMT1 in distinct cellular populations, thereby providing a systematic reference to facilitate novel clinical perspectives in the field of cardiovascular research. PRMTs, particular PRMT4/5, have effects on ECs that extend beyond the cardiovascular system and also impact peripheral circulations, primarily in the context of vascular regeneration following injury. PRMT4 has been found to perform restorative impacts in ischemic conditions. PRMT4 overexpressed in ischemic muscles as well as under vascular endothelial growth factor (VEGF) stimuli, promoting ECs angiogenic effects, manifested as elevated migration, proliferation, and tube formation, thereby culminating in boosting capillary density and hemodynamics recovery [39]. Mechanical investigations revealed the modification of PRMT4 in VEGF transcription and VEGF receptor 2 (VEGFR2) phosphorylation, supporting its essential position in maintaining ECs homeostasis. Consistent with PRMT4, PRMT5 expression was significantly upregulated in hypoxic ECs and ischemic tissues. Both genetic and pharmacological inhibition of PRMT5 impaired typical angiogenic features, accompanied by enhanced necrosis, in a mechanism of interfering hypoxia-induced factor 1-α and VEGF-related pathways in ECs [62]. Declined PRMT5 level was linked to oxidative stress and inflammation in ECs induced by oxidized low-density lipoprotein through interaction with programmed cell death protein 4, as well as overexpressed VCAM1 through impaired methylation of homeobox protein Hox-A9, respectively [63], collectively deteriorating ECs function. Further in-depth mechanistic investigations partially revealed the implication of PRMT5 in regulating inflammatory response and apoptosis in ECs. C-X-C motif chemokine 11 (CXCL11) is essential in atherosclerosis process, with a pro-inflammatory impact involved. The stimulation of CXCL11 in ECs relies on PRMT5-methylated NF-κB subunit p65 at Arg174. PRMT5 inhibition decreased the expression level of CXCL11 [34], emphasizing the significant effects of PRMT5 in EC inflammation. Researchers also found that PRMT5 functioned as a binding protein in ECs, methylating apoptosis signal-regulating kinase 1 (ASK1) at Arg89, a pivotal regulator of endothelial permeability and apoptosis, improving ASK1 phosphorylation at the Ser-83 site, ultimately preventing EC death [64].
Fibroblasts
Fibroblasts are critical in and function of cardiac tissue during CVDs, mainly in regulating fibrotic formation and contributing to the inflammatory response [65,66]. The regulatory role of PRMTs in fibroblasts within cardiovascular disorders has, however, received relatively little attention in recent years. Recent studies revealed a novel phenotype of the ablation of histidine decarboxylase, a well-recognized contributor of inflammatory response, promoted cardiac fibroblast proliferation and migration, accompanied with cardiomyocytes death, aggravating acute MI. Notably, PRMT1 emerged as a significant mediator in mediating these processes [67]. Another study elucidated the promotion of PRMT5 in fibrotic process. PRMT5 overexpression activated cardiac fibroblasts via transforming growth factor β1 in vitro and exacerbated adriamycin-induced cardiac fibrosis in vivo, whereas PRMT5 knockdown performed mitigating effects [68]. Nevertheless, its role in physiological states and its involvement in other pathological processes within the realm of CVDs remain to be fully elucidated.
PRMTs IN METABOLIC DISEASES
The effects of PRMTs in metabolic diseases has garnered increasing attention in recent years, highlighting their multifaceted involvements in the regulation of metabolic processes (Fig. 3). PRMTs wield precise control over key cellular pathways, including those governing glucose metabolism and lipid homeostasis. Emerging research has revealed PRMTs dysregulation is intricately associated with the pathogenesis of multiple metabolic disorders, such as diabetes, obesity, fatty liver diseases, etc. [69,70].
PRMTs in diabetes
Dysregulations in glucose metabolism culminate in the manifestation of diabetes [71,72], while increasing evidence have underscored the link between the genesis of diabetes and epigenetic variations, where histone modifications and DNA methylation serve as valuable indicators [73]. Recent research has illuminated the profound significance of PRMT1 in maintaining glucose homeostasis during the developmental process. PRMT1 maintains pancreas homeostasis and promotes development through affecting neurogenin 3, thereby participating in glycemic control [74]. Meanwhile, PRMT1 activated gene subsets necessary for the development of pancreatic endocrine and exocrine cells by recruiting PRMT4 or methylating histone H4, which in turn contributes to glucose metabolism. Furthermore, gluconeogenesis, a vital biological process in the progression of diabetes, has been revealed to have close associations with several PRMTs. For instance, liver-specific PRMT1 deficiency significantly ameliorated diabetic hyperglycemia by reducing forkhead box protein O1 (FOXO1)-dependent hepatic gluconeogenesis [75]. On the contrary, gain-of-function of PRMT1 variant 2 activated gluconeogenesis in hepatocytes through the interactions with peroxisome proliferator-activated receptor-gamma coactivator 1α (PGC-1α), a transcriptional coactivator on gluconeogenesis, thereby increasing its activity through methylation modification [76]. PRMT4 expression level has been found to elevate in individual afflicted with type 2 diabetes mellitus (T2DM) [77]. Within the diabetic milieu, PRMT4 activity and expression become indispensable for the transcription of genes intricately implicated in glycogen metabolism [78]. Remarkedly, PRMT4 has been unequivocally demonstrated to engage in interactions with the cAMP responsive element-binding protein (CREB) to modulate gluconeogenesis [79]. PRMT4 also repressed glycolytic flux and glycolysis through the hypermethylation of glyceraldehyde-3-phosphate dehydrogenase at Arg234 [80]. In a study conducted under fasting conditions, it was revealed that PRMT5 stimulated CREB phosphorylation through the modulation of methylation, thus intricately fine-tuning the process of gluconeogenesis [81]. Meanwhile, PRMT5 has emerged as a potent enhancer of the Small Heterodimer Partner, a nuclear receptor profoundly involved in metabolic homeostasis [82,83]. This enhancement occurs through the inhibition of the expression of metabolic target genes across various tissues, orchestrating glucose and lipid levels within the body [84]. Another noteworthy study has explored the interactions between PRMT6 and CREB regulated transcription coactivator 2 (CRTC2), a coactivator of CREB, by mediating arginine methylation of CRTC2 to strengthen the association between CRTC2 and CREB, contributing to hepatic glucose control [85]. To complement these mechanisms, research in T2DM rodent models has indicated that PRMT1 mediated insulin signaling as well as exerted control over hepatic gluconeogenic genes expression, glucose metabolism, and insulin secretion [86]. In type 1 diabetes mellitus mice, the overexpression of PRMT4 has been shown to reduce fat pad weights and adipocyte sizes, thus promotes lipolysis [87]. On the other hand, PRMT5 has implicated in the transcription processes of insulin genes. Islet-specific PRMT5 KO declined insulin gene expression, and affected glucose-stimulated insulin secretion and glucose tolerance in vivo, likely via histone methylation-regulated chromatin remodeling [88]. In addition, our prior data has demonstrated that PRMT7 deficiency in mice led to elevated blood glucose levels, accompanied by impaired glucose and insulin sensitivity. Remarkably, this deficiency does not impact gluconeogenesis, energy intake, or levels of leptin or insulin [89].
In addition, PRMTs are intricately associated not only with the pathogenesis of diabetes but also with its complications, including diabetic retinopathy and diabetic nephropathy. PRMT1 and dimethylarginine dimethylaminohydrolases (DDAHs)-induced ADMA upregulation have been identified as key implications of renin-angiotensin system- and ROS-mediated diabetic retinopathy [90]. Besides, ROS induced apoptosis to retinal pigment epithelial cells, via PRMT1 in a sirtuin 1 (SIRT1)-dependent manner [91] as well as PRMT4-dependent H3R17 dimethylation [92], thus triggering diabetic retinopathy. In the domain of diabetic nephropathy, PRMT1 essentially regulated the apoptosis and epithelial-mesenchymal transition in renal tubular epithelial cells through the activation of ER stress pathways, hence leading to diabetic nephropathy [93]. PRMT4 downregulation in response to high glucose levels is mediated by ubiquitination-dependent PRMT4 degradation, ultimately inducing podocyte loss and resulting in diabetic nephropathy [94].
Epigenetic modulation is imperative for β-cell maturation and identity maintenance. As a representative mechanism, histone methylation was proved to be essential for modulating β-cells functions. PRMT1-defiency in fetal and adult β-cells caused diabetic features, which was aggravated by metabolic stresses [95]. PRMT1 ablation in adult β-cells led to a rapid impairment of H4R3me2a and the ensuing loss of β-cell characteristics. However, in contrast, elevated PRMT1 expression was measured in individuals afflicted with diabetes [95,96], especially in β-cells exposed to high glucose. Silencing PRMT1 attenuated β-cell dysfunction, such as suppressed glucose-stimulated insulin secretion caused by high glucose treatment. This effect might be explained by the promotive effects of PRMT1 in glucose toxicity-stimulated β-cell dysfunction in a FOXO1-dependent manner [97], thus inducing hyperglycemia both in normoxic and hypobaric hypoxia condition [98]. In addition to PRMT1, PRMT4-dependent H3R17me2a in pancreatic β-cells was also detected to affect glucose-stimulated insulin secretion [99].
PRMTs in obesity
Obesity poses a myriad of deleterious consequences, amplifying the susceptibility to multiple disorders [71,100]. Indeed, the pivotal elements governing obesity, particularly adipogenesis and lipid metabolism [101], have recently been revealed to be intricately associated with PRMTs. Several key factors gaining certain attention in this context include CCAAT/enhancer-binding protein β (C/EBP-β) and peroxisome proliferator-activated receptor-γ (PPARγ). Depletion of PRMT1 exerted an inhibitory effect on adipogenesis, attenuating the expression of adipogenic genes, whereas PRMT1 overexpression elicited the converse response [102]. Mechanical study revealed that PRMT1 promoted the expression of PPARγ by catalyzing histone modification on H4R3me2a to accelerate adipogenic differentiation. Moreover, PRMT1 acted as an activator in preserving C/EBP-β via ameliorating the abundance of E3 ubiquitin ligase Smurf2. This decline impeded the degradation and ubiquitination of C/EBP-β, thus promoting adipogenic process. Concomitantly, promoter-enhancer looping at the PPARγ2 locus, a pivotal event in adipogenic differentiation, hinges upon PRMT5 activity [103]. In contrast, PRMT6 negatively regulated the activity of PPARγ by inhibiting its transactivity via the generation of a repressive epigenetic mark H3R2me2a [104]. In our prior studies, we reported a suppressive influence of PRMT7 in adipogenesis through C/EBP-β regulation [105]. Intriguingly, PRMT7 was also proved to be dispensable for adipogenic differentiation in tissue culture models [106]. Apart from these well-recognized factors, current research also revealed the role of PRMTs in the complex pathophysiology of obesity in general. PRMT1, for instance, was recognized to partake in inflammation-mediated hepatic lipogenesis, mainly through PGC-1α modulation [107]. PRMT2 functioned as a regulator of energy homeostasis via hypothalamic leptin-signal transducer and activator of transcription 3 (STAT3) signaling pathway, involving feeding, obesity, and energy metabolism through this process [108]. PRMT4, on the other hand, regulated adipose development and exerted control over adipogenic transcripts [109]. PRMT5 activated the transcription of genes related with adipogenic differentiation [110]. Our prior data indicated that PRMT7 deficiency in mice reduced PGC-1α expression, manifesting as declined exercise endurance and oxidative metabolism. PRMT7 KO mice exhibited aging obesity with accumulated body fat and enlarged white adipocytes, along with an increase in lipid droplet deposit in brown adipocytes [89]. Remarkably, our finding further indicated that the shift in muscle fiber types observed in the skeletal muscles of PRMT7-deficiency mice, characterized by a decrease in oxidative fiber types I and IIa, and an increase in glycolytic fiber types IIx and IIb, may also contribute to the obesity phenotype in these mice.
Adipocytes rely significantly on arginine methylation, by regulating gene expression and contributing to adipocyte differentiation and lipid metabolism. PRMT1 abundantly expressed in the white adipose tissues (WAT), triggered either by obesity in human or upon a high-fat diet in mouse models [111]. Adipocyte-specific PRMT1 depletion led to reduced fat mass. Notably, the depletion of PRMT1 induced PRMT6 overexpression and subsequently activated the transcriptional activity of FOXO3 in WAT. This, in turn, triggered the AMP-activated protein kinase (AMPK) cascade, corresponding to the enhanced lipophagy, mitochondrial lipid catabolism, and a consequent shrunken in lipid droplet within WAT. While PRMT1 adipocyte-specific KO mice exhibited increased adipose tissue inflammation, which impaired insulin tolerance. PRMT4, on the other hand, has been shown to enhance PPARγ-mediated gene transcription, promoting adipocyte differentiation [109]. Recent research has unveiled a more specific mechanism by which PRMT4 regulates PPARγ. It was found that PRMT4 methylates PPARγ at Arg240, initiating browning and thermogenesis in adipose tissue [112]. Adipocyte-specific PRMT5 KO mice demonstrated suppression in lipid droplet biogenesis and fatty acid metabolism in WAT, giving rise to lipodystrophy [113]. Our investigations also indicated that preadipocyte-specific PRMT7 depletion or PRMT7 ablation in mouse embryonic fibroblasts enhanced adipogenesis by promoting mitotic clonal expansion, an initial step of adipogenesis, whereas PRMT7 overexpression ameliorated these processes [105].
PRMTs in hepatic steatosis
Hepatic steatosis, also known as fatty liver disease, is marked by the excessive accumulation of fat within liver cells. While the research regarding the role of PRMTs in the pathogenesis of hepatic steatosis is still in its infancy, there are emerging indications that PRMTs indeed participate in and intervene with this pathological process. As exemplified, PRMT1 has been identified in the livers of obese individuals and in diet-induced obesity, where it exerted a protective effect by enhancing fatty acid oxidation and mitigating steatosis through PGC-1α [114,115]. PRMT1 prevented alcohol-induced liver damage via moderating oxidative stress process [116]. Contrarily, liver-specific PRMT1 depletion promoted the development of alcohol-stimulated hepatic steatosis via interfering hepatocyte nuclear factor 4 (HNF4) expression [117]. PRMT3 exhibited elevated expression in nonalcoholic fatty liver disease and interacted with liver X receptor α to enhance its activity, consequently promoting hepatic lipogenesis [26,118]. While PRMT3 inhibition has been shown to alleviate the severity of hepatic steatosis and reduce plasma lipid level [119].
Despite the substantial advancements in recent research, the investigation of PRMTs in metabolic diseases remains comparatively limited compared to their well-established roles in CVDs or cancers, warranting great demand of more dedicated research efforts. A thorough comprehension of PRMTs-mediated mechanisms in metabolic diseases holds immense promise for advancing our understanding and identifying innovative therapeutic strategies to address these prevalent health challenges.
PERSPECTIVES
Despite the limited extent of research conducted on PRMTs in cardiovascular system and metabolic disorders, accumulating evidence strongly indicates their indispensability in the pathogenesis and progression of these maladies, where PRMTs perform distinct roles in diverse cell types in terms of both physiological and pathological status. The precise modulation of PRMTs potentially represents novel strategies for the clinical treatment of diverse relevant diseases. Current ongoing efforts are dedicated to developing candidates targeting PRMTs for preclinical investigations. However, hitherto, only limited drugs, primarily inhibitors for PRMT5, have progressed into clinical trial phases (Table 1), with a predominant focus on antitumor research rather than cardiovascular or metabolic diseases [120]. The candidates, such as JNJ-64619178 and GSK3326595, have demonstrated promising preliminary efficacy, particularly in solid tumors with DNA damage repair deficiencies and splicing mutations [120-122]. Based on the extant clinical trials, there remains an anticipation for the acquisition of substantive evidence concerning the safety and efficacy of these agents within the human organism. This holds significant implications for guiding future endeavors in pharmaceutical design and clinical trials aimed at PRMTs within the context of cardiovascular and metabolic diseases. For instance, elevated PRMT5 expression triggered multiple pathological processes in specific tissues, such as atherosclerosis, myocardial fibrosis, gluconeogenesis, adipogenesis, and lipodystrophy, which enhanced the risk of various diseases including CVDs, diabetes, and obesity (Fig. 3). In this regard, PRMT5 inhibitors may provide potential benefits in modulating these pathophysiological progressions, thereby functioning as promising therapeutic interventions to various diseases such as CVDs, diabetes, and obesity. Nevertheless, considering the intricated roles of PRMT5 in distinct tissues, its inhibitors might also lead to certain unexpected pathological alterations, such as DCM and glucose tolerance impairment, further systematic evaluation and preclinical investigations are warranted. Meanwhile, it is imperative to recognize that numerous mysteries surround the current comprehension of PRMTs in these disorders, emphasizing great demands for continued research to unravel their intricate mechanisms.
In this review, we have synthesized current research findings to delineate promising avenues for therapeutic interventions targeting PRMTs. Understanding the regulatory effects of PRMTs in specific cell type may hold profound significance for delving deeper into the underlying mechanisms and shaping future therapeutic approaches. While considering that both overexpression and inhibition of PRMTs within cells may potentially impact cellular homeostasis and its responsiveness to stimuli, precise modulation of PRMTs under specific conditions emerges as an indispensable prerequisite for the efficacy and safety of therapeutic strategies. As such, further investigations delved into the specific roles of PRMTs in cardiovascular and metabolic diseases hold immense clinical significance, which may provide the promise of breakthroughs in understanding the underlying pathophysiology and paving the way for innovative therapeutic interventions.
 XML Download
XML Download