

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Cancer cachexia is a multifactorial syndrome characterized by muscle wasting that is irreversible by conventional nutritional support and negatively affects cancer patients’ quality of life [12]. Muscle wasting syndrome or muscle atrophy is induced by an imbalance between muscle protein degradation and synthesis [34]. Two E3 ubiquitin ligases, muscle RING-finger containing protein-1 (MuRF1) and muscle atrophy F-box protein, are muscle specific ligases that are upregulated in atrophying skeletal muscle and mediate the degradation of muscle protein [5]. Observations from recent studies that the knockdown of atrogin-1 or MuRF1 alleviates muscle atrophy in cancer cachexia implicated these 2 ligases as potential molecular targets for treating cancer cachexia-induced muscle atrophy [67]. During muscle atrophy, impaired amino acid delivery to skeletal muscles reduces muscle protein synthesis [8]. Therefore, amino acid starvation mechanisms within skeletal muscle should be inhibited to maintain skeletal muscle quality and strength [8].
The umami taste receptor (TAS1R1/TAS1R3) is expressed endogenously in various organ systems, including skeletal muscle tissues, and acts as an amino acid sensor [9]. TAS1R1/TAS1R3 expression has been reported to be decreased when autophagy is increased and is essential for amino acid-induced calcium influx [1011]. TAS1R1/TAS1R3 is also reported to be involved in myogenic differentiation in C2C12 cells; myogenic regulatory factors such as myoblast determination protein 1 (MyoD1) and myogenin regulate TAS1R3 [12], and MyoD1 upregulates Tas1r1 expression during differentiation [13].
Recently, TAS1R1/TAS1R3 was proposed to be involved in skeletal muscle metabolism and autophagy- related skeletal muscle diseases [911]. However, an endogenous mechanism of the umami taste receptor affecting skeletal muscle in muscle atrophy during cancer cachexia remains poorly elucidated.
Studies of the umami taste receptor have been less active than other types of taste receptors, such as sweet and bitter taste receptors, and little is known about the association between the umami taste receptor and any specific diseases. Given that the umami taste receptor is expressed endogenously in skeletal muscle and interacts with amino acids, it may be involved in mechanisms related to muscle wasting in skeletal muscle. Therefore, the present study investigated therapeutic potential of TAS1R1/TAS1R3 for muscle wasting in cancer cachexia and its mechanism of action in vivo and in vitro.
MATERIALS AND METHODS
In vivo experiments
Cancer cachexia mouse model
Five-week-old C57BL/6J male mice were obtained from Central Lab Animal, Inc. (Seoul, Korea). After a 1-week acclimation period, mice were randomly divided into 2 groups: a normal control mice group (CTRL; n = 6) and a Lewis lung carcinoma (LLC)-induced cancer cachexia mice group (CC; n = 6). Cancer cachexia was induced by using LLC cells (1 × 106) suspended in 100 μL phosphate-buffered saline, subcutaneously injected in the right hindlimb region of mice.
The tumor volumes were monitored every 2 days, and grip strength was measured weekly throughout the experiment. The volume of each tumor was measured using a digital caliper (BLUE BIRD, Seoul, Korea) and calculated with the following formula: [width (mm) × length × 0.52] [14]. Grip strength was measured using a grip strength meter (Jeungdo Bio & Plant, Seoul, Korea). To measure the grip strength of the mice, they were allowed to grasp the grid with all 4 paws, and their weight was sustained. Then, the mouse tail was gently pulled back horizontally until their grip was released from the grid was recorded as the peak force. Each mouse underwent 3 trials at a 1-min interval, and then the average was calculated. Grip strength was measured once a week.
A total of 21 days after tumor inoculation, the mice were anesthetized with isoflurane (Piramal Critical Care, Bethlehem, PA, USA). The tongue, hypothalamus, liver, duodenum, quadriceps muscle, triceps muscle, pectoralis muscle, gastrocnemius muscle, and tibialis anterior muscle were dissected, respectively, weighed, and stored at −80°C for further polymerase chain reaction (PCR) experiments.
The use of all mice was approved by the Institutional Animal Care and Use Committee (IACUC) of Ewha Womans University (IACUC No: 21-003-01), and all procedures were in accordance with the ARRIVE guidelines and the National Research Council’s Guide for the Care and Use of Laboratory Animals.
Histological analysis
Quadriceps muscle tissues were fixed in 10% formaldehyde. Paraffin-embedded tissues were cut into 5 μm sections and stained with hematoxylin and eosin. Three fields per muscle tissue and 50 fibers per field were examined under a light microscope (Nikon, Tokyo, Japan), and the muscle fiber cross-sectional area (μm2) was measured by tracing structures of muscle fibers for each mouse (n = 3/group) using ImageJ software (NIH, Bethesda, MD, USA).
In vitro experiments
Cell culture and reagents
C2C12 myoblasts and LLC cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA), and human skeletal myoblasts (HSkM) were purchased from Gibco (Thermo Fisher Scientific, Seoul, Korea). Cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Welgene, Daegu, Korea) supplemented with 10% fetal bovine serum (Gibco, Waltham, MA, USA) and 1% penicillin/streptomycin (100 U/mL and 100 μg/mL) (Invitrogen, Carlsbad, CA, USA). Cells were kept in a humidified incubator at 37°C and 5% CO2.
Preparation of LLC conditioned media
When the LLC cells reached > 90% confluency, the growth medium was removed. Conditioned medium (CM) was collected after 48 h incubation in serum-free DMEM, centrifuged to remove cell debris, and filtered using a syringe filter with a 0.2 μm pore size.
Myoblast differentiation and LLC CM treatment
C2C12 myoblasts and HSkM were grown to 85–90% confluence and were differentiated into myotubes by culturing the cells for 96 h (C2C12 myoblasts) and 48 h (HSkM) in DMEM (Welgene) with 2% horse serum (Gibco) and 1% penicillin. To establish cancer cachexia condition in vitro, fully differentiated myoblasts were incubated for 48 h a 1:2 ratio of LLC CM and fresh DMEM supplemented with 2% horse serum and 1% penicillin, replacing the medium every 24 h. Fully differentiated cells were set as a control, the CTRL group. Fully differentiated cells treated with LLC CM were set as the LLC-induced cancer cachexia, the CC group.
Transient transfection and treatment
HSkM were fully differentiated, then replaced with either fresh DMEM containing 2% horse serum for the CTRL group or LLC CM with 2% horse serum for the CC group prior to transient transfection. The cells were transfected with the mammalian expression vector pCMV6-ENTRY containing human TAS1R1 using Lipofectamine3000 (Invitrogen) mixed with Opti-MEM (Thermo Scientific, Waltham, MA, USA) according to the manufacturer's protocol [15]. All transfected cells were cultured for 48 h, and pCMV6 (empty vector [EV]) was used as a control.
RNA interference and treatment
Pooled siRNA (ON TARGETplus Human TAS1R1 siRNA) and control siRNA (ON-TARGETplus Non-targeting Control Pool) were purchased from Dharmacon (Lafayette, CO, USA). siRNA transfection in HSkM was performed using DharmaFECT1 reagent (Dharmacon) according to the protocols provided by the manufacturers. HSkM were fully differentiated, then replaced with either fresh DMEM containing 2% horse serum for the CTRL group or LLC CM with 2% horse serum for the CC group prior to siRNA transfection. All transfected cells were cultured for 48 h.
In vivo and in vitro experiments
RNA isolation and reverse transcription-quantitative PCR (RT-qPCR) analysis
RNA isolation was performed using TRIzol reagent (Invitrogen), and the RNA concentration and quality were checked using Nanodrop (Thermo Scientific). DNA was synthesized with RevertAid reverse transcriptase (Thermo Scientific) and quantitative PCR (qPCR) was performed using Rotor-Gene SYBR Green PCR Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. β-actin was used as a loading control for in vivo studies and in vitro C2C12 cell studies, and GAPDH was used for in vitro HSkM cell studies. The qPCR was performed using the following sequences for the forward (F) and reverse (R) primers: mouse F-Tas1r1, 5’-CATCTGGTGATTCTTGAGTG-3’ and R-Tas1r1, 5’-AGGATACGAAGTGGAGGAG-3’; mouse F-Tas1r3, 5’-CAGGCAGTT GTGACTCTGT TG-3’ and R-Tas1r3, 5’- TGCGATGCAGATACCTCGTG-3’; mouse F-Atrogin-1, 5’- AGTGAGGACCGGCTACTGTG and R-Atrogin-1, 5’- GATCAAACGCTTGC GAATCT-3’; mouse F-MuRF1, 5’- TGACATCTACAAGCAGGAGTGC-3’ and R-MuRF1, 5’- TCGTCTT CGTGTTCCTTGC-3’; mouse F-β-actin, 5’-CGCCACCAGTTCGCCATGGA-3’ and R-β-actin, 5’-TACAGCCCGGGGAGCATAGT-3’; human F-TAS1R1, 5’- GTTCTGCC TACAACGCATAC-3’ and R-TAS1R1, 5’- GTGTAGAAGGAAATGCACCTT-3’; human F-TAS1R1, 5’- GACAGAGCGCCTGAAGAT-3’ and R-TAS1R3, 5’- AGTCCACACAGTC GTAGCAG-3’; human F-Atrogin-1, 5’- TCACAGCTCACATCCCTGAG-3’ and R-Atrogin-1, 5’- AGACTTGCCGACTCTTTGGA-3’; human F-MuRF1, 5’- TGGACTTCTTTACTTT GG ATTTAG-3’ and R-MuRF1, 5’- CTTCCTCTTCCTGATCTTCTTC-3’; human F- β-actin, 5’- GATCATTGCTCCTCCTGAGC-3’ and R-β-actin, 5’- ACTCCTGCTTGCTGATCCAC-3’; human F-GAPDH, 5’-GACAGTCAGCCGCATCTTCT-3’ and R-GAPDH, 5’-GCGCCCAATACGACCAAATC-3’.
Western blot assay
Whole-cell proteins were extracted using a RIPA lysis buffer (50 mM Tris-Cl [pH 7.5], 150 mM NaCl, 0.1% sodium dodecyl sulfate [SDS], 0.5% sodium deoxycholate, 1% Nonidet P40, 1 mM NaF, 1 mM Na3VO4 and 1 mM PMSF) added with a protease inhibitor cocktail (Sigma-Aldrich, St. Louis, MO, USA). To quantify the protein concentrations, a protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA) was used, and 10–30 µg of protein samples were separated using SDS-polyacrylamide gel electrophoresis. All separated proteins were electro-transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA), blocked with 5% skimmed milk or bovine serum albumin in Tris-buffered saline containing Tween 20 (TBS-T), and probed with diluted primary antibodies, including Atrogin-1 (Santa Cruz Biotechnology, Santa Cruz, CA, USA, sc-166806), MYH (Santa Cruz Biotechnology, sc-376157), MuRF1 (Invitrogen, PA5-76695), Phospho-Akt (Ser473) (Cell Signaling Technology, Beverly, MA, USA, 9271), Akt (Cell signaling Technology, USA, 9272), Phospho-PI3K p85 (Tyr458)/p55 (Tyr199) (Cell signaling Technology, USA, 4228), PI3K (Cell Signaling Technology, USA, 4292), and α-tubulin (Abcam, Cambridge, UK, ab7291). Then, the membranes were washed with TBS-T and incubated with anti-mouse (Abcam, ab6708) or anti-rabbit secondary antibody (Santa Cruz Biotechnology, sc-2357) for 1 h at room temperature. The bands were visualized with a chemiluminescence kit (Advansta, Menlo Park, CA, USA), and the intensity of the bands was quantified with the ImageJ software (NIH).
Statistical analysis
All data are presented as mean ± SEM from at least 3 independent experiments. One-way analysis of variance, followed by Newman–Keuls' post hoc tests, was used for comparing 3 or more samples. For all other statistical analyses, a 2-tailed Student’s t-test was utilized. A P-value less than 0.05 was considered statistically significant. GraphPad PRISM (GraphPad Software, San Diego, CA, USA) was used for statistical analysis.
RESULTS
Muscle atrophic features in LLC-induced cancer cachexia mouse model
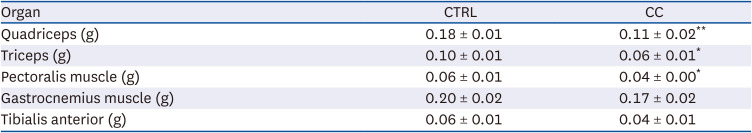
The cancer cachexia mouse model was established using LLC cells. The tumor volume gradually increased after inoculation of LLC cells and showed a significant difference from the CTRL group from the 12th day after injection (Fig. 1A). After tumor removal, carcass weight was significantly reduced by 13% in the CC group compared to the CTRL group (Fig. 1B). Significant decreases in quadriceps, triceps, and pectoralis muscle weights of the CC group were observed compared to the CTRL group (Table 1). In particular, the quadriceps of the CC group weighed 20% less than those of the CTRL group. To evaluate muscle function, grip strength was tested (Fig. 1C). There was a significant loss of grip strength in the CC group from 1 week after LLC cell inoculation.
Fig. 1
Muscle atrophic features in LLC-induced cancer cachexia mouse model. (A) Tumor volumes, (B) carcass weights after tumor removal (carcass-tumor weight), and (C) grip strength were measured (n = 6/group). (D) The representative quadriceps tissues stained with H&E were shown (magnification 100×, scale bar 100 μm), and (E) their myofiber size was quantified (n = 3/group). mRNA expressions of muscle atrophy-related genes (F) Atrogin-1 and (G) MuRF1 in quadriceps, triceps, and pectoralis muscle were determined by RT-qPCR (n = 6/group). All data were shown as mean ± SEM and analyzed by an unpaired 2-tailed Student’s t-test.
CTRL, control; CC, LLC-induced cancer cachexia; H&E, hematoxylin and eosin.
P < 0.05 was considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001).
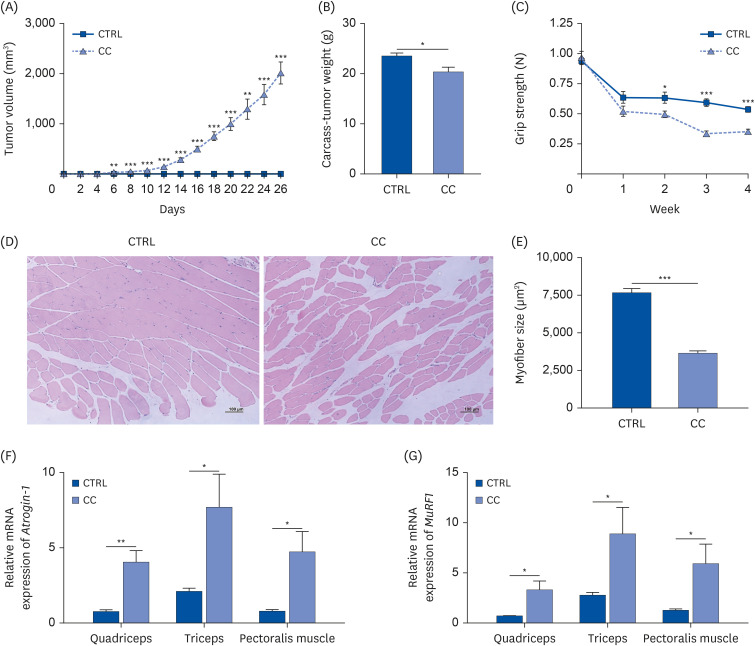
Table 1
Muscle weights of mice in LLC-induced cancer cachexia mouse model
To confirm whether muscle atrophy was sufficiently induced in the cancer cachexia model, the cross-sectional area of the quadriceps muscle was compared between the 2 groups. Muscle fiber size of the quadriceps muscle was significantly smaller in the CC group than that in the CTRL group, indicating that muscle atrophy had occurred (Fig. 1D and E). Moreover, the mRNA levels of atrophy-related markers, Atrogin-1 and MuRF1 were upregulated in the CC group compared to the CTRL group in all the quadriceps, triceps, and pectoralis muscles (Fig. 1F and G).
Umami taste receptor is expressed in different types of skeletal muscle and is downregulated in cachexia conditions in in vivo mouse model
To observe the levels of the umami taste receptor in skeletal muscles compared to other organs, the mRNA expression of Tas1r1 and Tas1r3 was analyzed in mouse tongue, hypothalamus, liver, duodenum, quadriceps, triceps, pectoralis muscle, and gastrocnemius muscles of the CTRL group (Fig. 2A and B). Expression of both Tas1r1 and Tas1r3 was higher in all muscles than in the tongue, duodenum, and liver. Previous studies have shown that Tas1r1 and Tas1r3 are widely expressed in various types of mouse tissues, and in particular, Tas1r1 is relatively highly expressed in muscle tissue compared to other tissues [9]. Contrary to the fact that taste receptors are mainly expressed in the tongue, in the present study, Tas1r1 expression was significantly higher in triceps (P < 0.05) and pectoralis muscle (P < 0.001), and Tas1r3 expression was significantly higher in triceps (P < 0.001), pectoralis muscle (P < 0.001), and gastrocnemius muscle (P < 0.05) than in the tongue, suggesting the possible physiological role of this taste receptor in skeletal muscle.
Fig. 2
Umami taste receptor is expressed in different types of skeletal muscle and is downregulated in cachexia condition in an in vivo mouse model. The mRNA expressions level of (A) Tas1r1 and (B) Tas1r3 were analyzed in mouse tongue, hypothalamus, liver, duodenum, quadriceps, triceps, pectoralis muscle, and gastrocnemius muscles of the CTRL group (n = 6/group). The mRNA expressions of (C) Tas1r1 and (D) Tas1r3 in quadriceps, triceps, and pectoralis muscles were analyzed. β-actin was used as a loading control (n = 6/group). Data were analyzed by one-way analysis of variance with a Newman-Keuls’s post hoc test. Different letters indicate statistical significance (P < 0.05), and an unpaired 2-tailed Student’s t-test was performed to compare data between 2 groups. All bars represent the mean ± SEM.
CTRL, control; CC, LLC-induced cancer cachexia.
P < 0.05 was considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001).
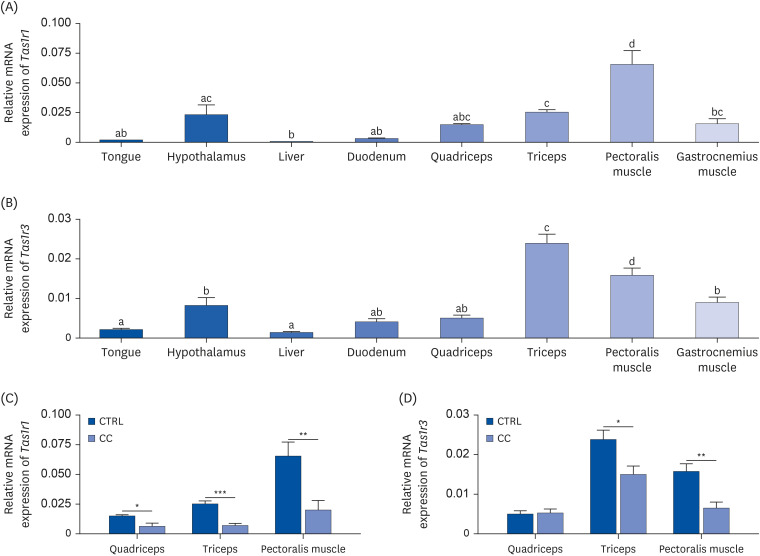
To investigate whether there is a change in umami taste receptor expression due to a cancer cachexia state, differences in the receptor expression between the CTRL and CC groups were analyzed in 3 types of muscles. Tas1r1 expression in the quadriceps, triceps, and pectoralis muscles was decreased in the CC group compared to the CTRL group by 57% (P < 0.05), 72% (P < 0.001), and 69% (P < 0.01), respectively (Fig. 2C). Tas1r3 expression showed a significant decrease in triceps and pectoralis muscles, but not quadriceps, in the CC group compared to the CTRL group by 37% (P < 0.05) and 59% (P < 0.01), respectively (Fig. 2D). In addition, the significant downregulation of Tas1r1 in the CC group was observed only in muscle tissues. There was no change in the liver, and the tongue and hypothalamus tended to decrease without significance in the CC group compared to the CTRL group (data not shown). These results suggest that umami taste receptor is endogenously highly expressed in skeletal muscle and its expression levels are downregulated in cancer cachexia conditions.
Muscle atrophy markers are upregulated, and umami taste receptor is downregulated in cachexia conditions in in vitro mouse model
To confirm the expression of the umami taste receptor in a mouse in vitro system, cancer cachexia conditions were established using the mouse myoblast cells with LLC CM treatment. The induction of differentiation was verified by measuring myosin heavy chain (MYH) levels, a marker gene that distinguishes myogenic and non-myogenic differentiation (Supplementary Fig. 1). The mRNA and protein expressions of Atrogin-1 and MuRF1 were examined to confirm whether atrophy was properly induced in the CC group. Both of mRNA and protein expressions of Atrogin-1 and MuRF1 were significantly upregulated in the CC group compared to the CTRL group (Fig. 3A-E). Furthermore, to examine the PI3K/Akt pathway, which is related to muscle atrophy in vitro, the protein levels of phosphorylation of PI3K and Akt were examined by Western blot (Fig. 3F-H). The phosphorylation levels of PI3K and Akt were significantly decreased in the CC group compared to the CTRL group.
Fig. 3
Muscle atrophy markers are upregulated and the umami taste receptor is downregulated in cachexia conditions in an in vitro mouse model. The mRNA levels of (A) Atrogin-1 and (B) MuRF1 were analyzed by RT-qPCR. The protein levels of (C-E) Atrogin-1, MuRF1, and (F-H) PI3K/Akt pathway in C2C12 cells were analyzed by Western blotting. (C, F) Representative blots were shown. (D, E, G, H) The relative band intensities were calculated after normalization to α-tubulin expression. The mRNA levels of (I) Tas1r1 and (J) Tas1r3 were measured by RT-qPCR. All values were shown as mean ± SEM (n = 3). Data were analyzed by an unpaired 2-tailed Student’s t-test.
CTRL, control; CC, LLC-induced cancer cachexia; MuRF1, muscle RING-finger protein-1; RT-qPCR, reverse transcription-quantitative polymerase chain reaction; PI3K, phosphoinositide 3-kinase.
P < 0.05 was considered statistically significant (*P < 0.05, **P < 0.01).
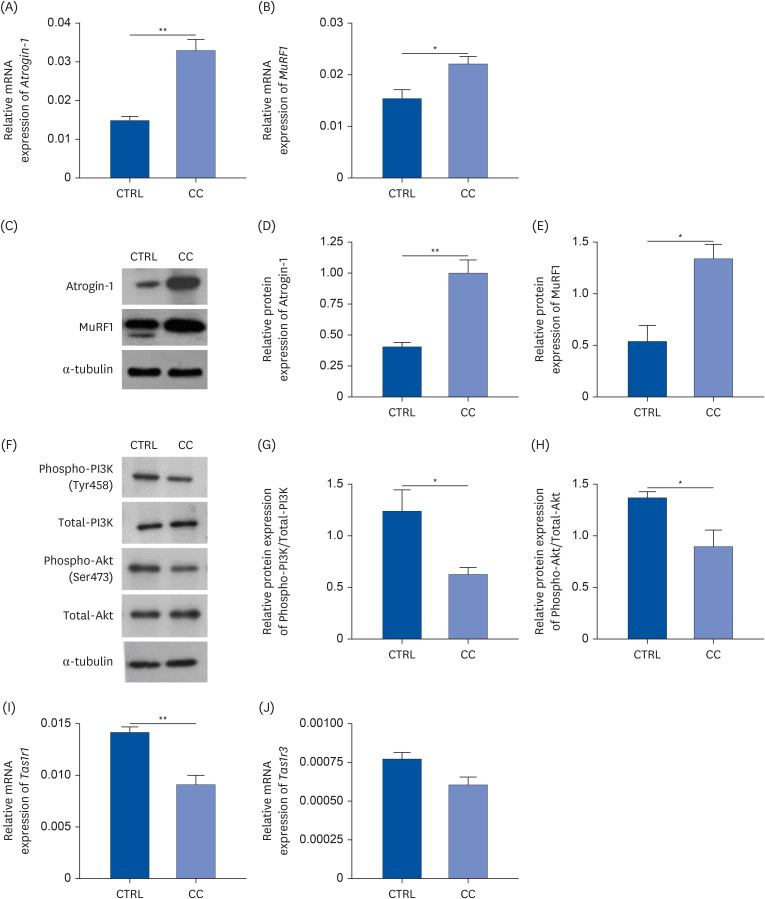
The mRNA levels of Tas1r1 and Tas1r3 were analyzed in the CTRL and CC groups and compared to confirm the downregulation of the umami taste receptor expression in vitro (Fig. 3I and J). Compared to the CTRL group, Tas1r1 expression was significantly downregulated in the CC group by 35% (P < 0.01), while Tas1r3 expression tended to be decreased without significance. These results were consistent with the in vivo results which showed decreased Tas1r1 expression in the CC group compared to the CTRL group.
Umami taste receptor is downregulated in in vitro human primary cells and exerts an anti-atrophic role
To confirm the umami taste receptor expression in vitro in a human primary cell model, HSkM cells were used. The mRNA levels of TAS1R1 and TAS1R3 were measured in the CTRL group and the CC group (Fig. 4A and B). Compared to the CTRL group, TAS1R1 expression was significantly downregulated in the CC group by 65% (P < 0.001), whereas there was no significant difference in TAS1R3 expression. These results were consistent with the mouse in vitro and in vivo experiments, suggesting that the umami taste receptor is downregulated in cachexia conditions and that TAS1R1 might play a major role in skeletal muscle regulates muscle atrophy in cancer cachexia.
Fig. 4
Umami taste receptor is downregulated in cachexia conditions in an in vitro human model and suppresses atrophic markers. The mRNA levels of (A) TAS1R1 and (B) TAS1R3 were analyzed by RT-qPCR. (C-E) Fully differentiated HSkM cells were transfected with pCMV6 EV or pCMV6-Entry containing TAS1R1. The mRNA levels of (C) TAS1R1, (D) Atrogin-1, and (E) MuRF1 were analyzed by RT-qPCR. (F-H) Fully differentiated HSkM cells were transfected with control siRNA or TAS1R1 siRNA. The mRNA levels of (F) TAS1R1, (G) Atrogin-1, and (H) MuRF1 were measured by RT-qPCR. GAPDH was used as a loading control and all experiment results are shown as mean ± SEM (n = 3). Data were analyzed by one-way analysis of variance with a Newman-Keuls’s post hoc test. Different letters indicate statistical significance (P < 0.05), and an unpaired 2-tailed Student’s t-test was performed to compare data between 2 groups.
CTRL, control; CC, LLC-induced cancer cachexia; EV Ctrl, empty vector control; CTRL + EV, control + empty vector; CTRL + TAS1R1, control + TAS1R1; CC + EV, LLC-induced cancer cachexia + empty vector; CC + TAS1R1, LLC-induced cancer cachexia + TAS1R1; non, non-targeting siRNA; CTRL + non, control + non-targeting siRNA; CTRL + siTAS1R1, control + TAS1R1 siRNA; CC + non, LLC-induced cancer cachexia + non-targeting siRNA; CC + siTAS1R1, LLC-induced cancer cachexia + TAS1R1 siRNA; MuRF1, muscle RING-finger protein-1; RT-qPCR, reverse transcription-quantitative polymerase chain reaction.
P < 0.05 was considered statistically significant (**P < 0.01, ***P < 0.001).
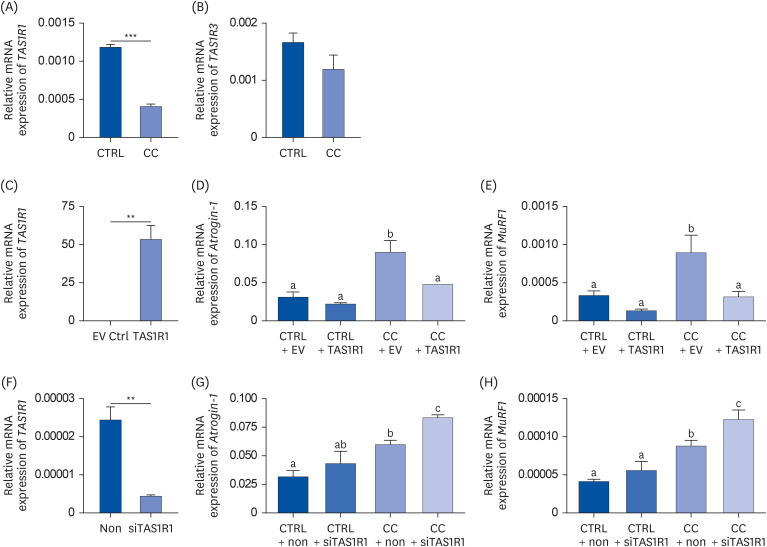
To determine whether atrophy can be restored by overexpressed TAS1R1, TAS1R1 was transiently transfected into fully differentiated HSkM cells. The overexpression efficiency was verified by RT-qPCR (Fig. 4C). Overexpressed TAS1R1 significantly downregulated Atrogin-1 and Murf1 in the CC + TAS1R1 group compared to the CC + EV group by 47% (P < 0.01) and 65% (P < 0.05), respectively, suggesting that TAS1R1 can prevent muscle atrophy in a muscle wasting condition (Fig. 4D and E). No significant difference was observed in the Atrogin-1 and Murf1 genes between the CTRL + EV and CTRL + TAS1R1 groups.
Moreover, to investigate whether the knockdown of TAS1R1 exacerbates muscle atrophy, fully differentiated HSkM cells were transfected with control siRNA or TAS1R1 siRNA for 48 h. The knockdown efficiency was verified by RT-qPCR (Fig. 4F). Downregulated TAS1R1 significantly upregulated Atrogin-1 and MuRF1 by 40% (P < 0.05) and 40% (P < 0.05) in the CC + siTAS1R1 group compared to the CC + non group (Fig. 4G and H). No significant difference was observed in 2 genes between CTRL + non and CTRL + siTAS1R1 groups. These results demonstrate that TAS1R1 blocks muscle atrophy in skeletal muscle under cancer cachexia conditions.
DISCUSSION
In this study, a novel role of the umami taste receptor under muscle wasting conditions was investigated. Expression of TAS1R1 was significantly downregulated in skeletal muscles during cancer cachexia, and the level of muscle atrophy was inversely correlated with the degree of TAS1R1 expression.
Of the 5 distinct types of taste receptors, umami, sweet, and bitter tastes are mediated by G protein-coupled receptors (GPCRs) for their sensation [16]. One type of taste GPCR, the taste receptor type 1 (TAS1R) family, consists of 3 subunits: TAS1R1, TAS1R2, and TAS1R3, which form heterodimers, such as TAS1R1/TAS1R2 for the sweet taste receptor and TAS1R1/TAS1R3 for the umami taste receptor [17]. Although TAS1Rs are generally known to be expressed in oral tissues, studies have shown that TAS1Rs are also expressed and function in extra-gustatory organs [18]. In addition, their expression and function are altered during disease states, which suggests that these taste receptors may be a therapeutic target for certain diseases [16]. Moreover, because GPCRs are the largest family of individual drug targets [19], it was hypothesized that the umami taste receptor in skeletal muscles might be a potential therapeutic target for treating muscle wasting. In the present study, the umami taste receptor expression was altered in skeletal muscles under cancer cachexia conditions. Having shown that the expression of TAS1R1 was significantly downregulated in the cancer-induced muscle wasting condition of cachexia, both in vivo and in vitro, we hypothesized that TAS1R1 might regulate muscle atrophy in skeletal muscle. It was confirmed that overexpression of TAS1R1 restored muscle atrophy in the CC group. Furthermore, the knockdown of TAS1R1 exacerbated muscle atrophy in the HSkM cells of the CTRL group and the CC group.
It remains to be investigated whether the atrophic regulatory function of TAS1R1 in the skeletal muscle is a solitary effect or whether TAS1R3 is also involved. However, the distinct ligand specificity of TAS1R1/TAS1R3 and TAS1R2/TAS1R3 suggests that TAS1R1 and TAS1R2 are mainly involved in ligand binding in sweet and umami taste receptors disproportionately more than TAS1R3 [20]. Hence, we speculate that TAS1R1 can be a representative receptor for the umami taste receptor, but because the umami taste receptor is a heteromeric complex, the functionality of TAS1R3 in skeletal muscle may not be excluded.
Previously, the highest expression of Tas1r1 was shown in muscle compared to all other tissues [9], which was consistent with our results, suggesting its important role in muscle. Unlike the previous study which analyzed Tas1r1 expression in 1 representative muscle, the expression was analyzed in 4 different types of muscle in the present study. We provided the different expression depending on the type of muscle among the quadriceps, triceps, pectoralis muscle, and gastrocnemius muscle. Further research is needed to determine whether TAS1R1/TAS1R3 affect muscle atrophy differently according to different muscle types.
Multiple signaling pathways are associated with skeletal muscle growth and atrophy by regulating protein turnover in muscle tissues [21]. As a treatment for cancer cachexia, recent studies have focused on the protective effects against muscle wasting by downregulating the main pathways involved in muscle atrophy. The activation of the PI3K/Akt pathway is downregulated during muscle atrophy, which results in downstream activation of FoxO3, a target of the E3 ubiquitin ligases [2223]. In this context, our findings suggest that the umami taste receptor plays a role in gene regulation involved in muscle atrophy by regulating PI3K/Akt pathways. In addition, it has been reported that autophagy has been activated in muscle atrophy conditions, including cancer cachexia [24]. TAS1R1/TAS1R3 has been down-regulated when autophagy is activated [10]. This autophagy was regulated by the PI3K/Akt pathway [25]. In the present study, TAS1R1/TAS1R3 expression has decreased in the triceps and pectoralis muscle in the cancer cachexia model and PI3K/Akt phosphorylation was inhibited in the cancer cachexia model. Therefore, one of the mechanisms of this taste receptor in anti-muscle atrophy may be the inhibition of autophagy in the muscle by activating the PI3K/Akt pathway. This will be investigated in future studies.
In this study, mouse in vivo and in vitro models and an in vitro human primary cell model were used to compare the expression of the umami taste receptor under cachectic conditions. TAS1R1 was significantly downregulated under cachectic conditions in all 3 models, whereas TAS1R3 showed an insignificant decrease in the CC group in both in vitro models compared to the CTRL group. In addition, the mRNA expression of Tas1r3 showed a significant decrease in the triceps and pectoralis muscle in the mouse in vivo model, whereas there was no significant difference in the quadriceps between the groups. Although muscle atrophy occurred significantly in all 3 muscles, the levels of Tas1r3 expression between groups differed by muscle type. Moreover, when the Tas1r3 mRNA expression was analyzed in C2C12 cells, the gene expression was not consistent (data not shown). This may be caused by the limited in vitro models representing various types of skeletal muscle in mice.
The strength of this study is that the novel role of the umami taste receptor in skeletal muscle wasting was found for the first time. Although the umami taste receptor is reported to be expressed in skeletal muscle [9], its pathophysiological function in the muscle is not yet fully understood. Our study found that TAS1R1 is significantly suppressed in skeletal muscle during cancer cachexia in mice in vitro and in vivo, and these results were confirmed using HSkM cells. This finding can be applied to other wasting diseases for therapeutic purposes and could be a new starting point to investigate additional roles and mechanisms of the umami taste receptor besides its role as a taste sensor. However, since our findings are limited to measuring changes in mRNA levels only, further studies on protein changes should be conducted as mRNA may not represent protein changes.
Studies in the field of cancer cachexia research have shown that skeletal muscle loss is a common occurrence during cancer development and growth. The current study suggests that TAS1R1 may be able to preserve skeletal muscles during tumor growth. Another possible clinical implication of the present study is that combining umami taste substances that increase TAS1R1 expression with other treatments could be a possibility. Further study is needed to fully understand how TAS1R1 exerts therapeutic potential in other muscle-wasting conditions, such as sarcopenia, hypertension, and chronic heart failure. In conclusion, TAS1R1 exerts anti-atrophic effects in muscle wasting conditions, which suggests a therapeutic role for the umami taste receptor in cancer cachexia.
 XML Download
XML Download