

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
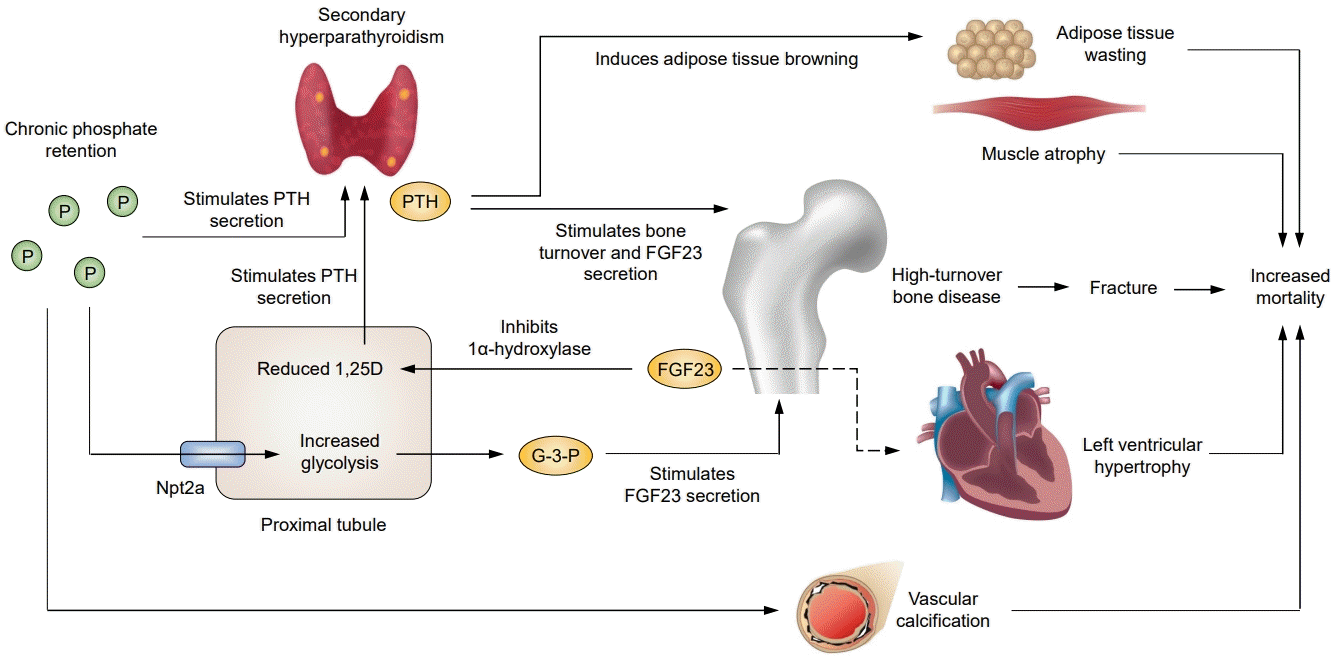
Patients with chronic kidney disease (CKD) experience a progressive deterioration of mineral metabolism, including conditions such as hyperphosphatemia and hypocalcemia, as their kidney function declines. Parathyroid hormone (PTH) and fibroblast growth factor 23 (FGF23) play important roles in the pathophysiology of mineral disturbances and subsequent bone abnormalities [1]. Several cohort studies have shown that fracture risk increases as kidney function deteriorates [2,3], suggesting the importance of therapeutic interventions for bone abnormalities in CKD patients. Observational studies have also shown an increased risk of cardiovascular disease and mortality in this population [4-6]. This increased risk is attributable not only to traditional factors but also to CKD-specific factors, such as mineral imbalances. Consequently, the term “CKD-mineral and bone disorder” (CKD-MBD) has been proposed to describe the broader clinical syndrome involving mineral metabolism, bone abnormalities, and vascular calcification [7]. PTH and FGF23 are important components of CKD-MBD and have been the focus of extensive research over the years.
This review provides an overview of the role of PTH and FGF23 in the pathophysiology of CKD-MBD. It also discusses the current understanding of relevant adverse outcomes of CKD-MBD and the management of this condition.
CHANGES IN PTH AND FGF23 WITH PROGRESSION OF CKD
CKD-MBD develops in the early stages of CKD. As kidney function declines, the number of functioning nephrons decreases and urinary phosphate excretion decreases. FGF23, which is secreted by osteocytes, increases phosphate excretion per nephron [8-10], presumably as a compensatory response. However, FGF23 suppresses the biosynthesis of 1,25-dihidryoxyvitamin D (1,25D) in proximal tubular cells, which decreases the intestinal absorption of calcium and phosphate. Consequently, PTH secretion in the parathyroid glands increases, which also increases phosphate excretion per nephron [11]. Thus, in patients with early-stage CKD, circulating levels of FGF23 and PTH increase to maintain a neutral phosphate balance by increasing urinary phosphate excretion, but as a trade-off, secondary hyperparathyroidism (SHPT) begins to progress. In patients with moderate to advanced CKD, decreased kidney expression of Klotho attenuates the ability of the kidney to excrete urinary phosphate, leading to an increase in serum phosphate. This process is accompanied by a progressive reduction in 1,25D levels through the inhibition of CYP27B1 by hyperphosphatemia [11], which further stimulates PTH secretion and causes serum calcium levels to decrease. In patients with advanced-stage CKD, the progression to kidney failure overcomes the compensatory system of FGF23 and PTH to maintain a neutral phosphate balance with a decreased nephron mass, resulting in overt hyperphosphatemia, hypocalcemia, and SHPT.
When patients reach kidney failure, the circulating levels of PTH and FGF23 increase 5- to 10-fold and 10- to 50-fold above normal, respectively. We prospectively studied the effect of hemodialysis initiation and found progressive reductions in serum PTH and FGF23 levels, with dialysis-related fluctuations [12]. We also found that the magnitude of FGF23 reduction was strongly associated with concomitant changes in serum phosphate levels, supporting the view that phosphate is a strong regulator of FGF23 production.
However, the underlying mechanism through which phosphate increases FGF23 production in osteocytes remains incompletely understood. Emerging evidence indicates that phosphate promotes glycolysis in proximal tubular cells via the type 2a sodium-dependent phosphate cotransporter. Consequently, kidney-derived glycerol-3-phosphate (G-3-P), a byproduct of glycolysis, stimulates the production of FGF23 by osteocytes [13]. The absence of an increase in G-3-P levels in the liver, heart, or skeletal muscle after phosphate loading suggests that phosphatestimulated G-3-P production is likely specific to the proximal tubules. In cases of acute kidney injury, levels of circulating G3-P derived from the injured kidney increase rapidly, thereby stimulating osteocytes to produce FGF23 [14]. However, limited information exists regarding the relationship between G-3-P and FGF23 in patients with CKD. Further studies are needed to elucidate the role of G-3-P in CKD.
CLINICAL MANIFESTATIONS OF SHPT
Bone abnormalities
Patients with advanced CKD develop various skeletal manifestations as SHPT progresses. The most common skeletal manifestation of severe SHPT is high-turnover bone disease (osteitis fibrosa), which is characterized by excessive rates of bone resorption and formation, increased cortical porosity, and progressive bone loss [15,16]. A longitudinal study of patients with CKD stages 2 to 5D demonstrated that a decrease in cortical bone loss over time was associated with elevated levels of PTH and bone turnover markers [17], suggesting that SHPT-mediated cortical porosity increases bone fragility and fracture risk. Inconsistent findings have been reported regarding the association between PTH levels and fracture risk, but the international Dialysis Outcomes and Practice Patterns Study (DOPPS) [18] and the European Current Management of Secondary Hyperparathyroidism: A Multicenter Observational Study (COSMOS) [19] have shown that intact PTH levels above 800 to 900 pg/mL are independently associated with an increased risk of fragility fractures.
Mortality associated with SHPT
Importantly, several cohort studies from different regions of the world have consistently shown that SHPT is associated not only with the risk of fragility fractures, but also with mortality [20-23]. Of note, a recently published large prospective cohort study using data from the nationwide registry of Japanese dialysis patients showed that higher intact PTH levels were almost linearly associated with an increased risk of all-cause and cardiovascular mortality in a time-averaged model [23]. We and others have also reported significant associations between parathyroidectomy and improved survival [24-26]. Although these studies are observational and cannot prove causality, the associations remained significant after adjustment for potential confounders, suggesting that intensive PTH control, as seen after parathyroidectomy, may contribute to better clinical outcomes in patients with kidney failure.
POTENTIAL MECHANISMS LINKING SHPT AND MORTALITY IN ADVANCED CKD
To support the validity of therapy aiming to control PTH, it is important to elucidate the mechanisms by which elevated PTH might lead to poor clinical outcomes. Fractures and vascular calcification are often considered as possible mechanisms, but these cannot fully explain the association between SHPT and increased mortality. In this context, an emerging potential mechanism is wasting, which is characterized by increased energy expenditure. A previous clinical study of hemodialysis patients reported that elevated PTH levels were an independent determinant of increased energy expenditure, as measured by indirect calorimetry [27]. Moreover, the increased energy expenditure diminished significantly in all patients 6 months after parathyroidectomy, suggesting that PTH plays a key role in the pathogenesis of wasting in advanced CKD. The mechanisms underlying these observations have been elucidated through insights from cancer research. Kir et al. [28] discovered that PTH-related protein (PTHrP), a tumor-derived protein that causes hypercalcemia of malignancy, directly drives thermogenesis in adipose tissue by switching from white to brown adipocytes (a phenomenon termed adipose tissue browning), suggesting that PTHrP mediates energy wasting in adipose tissue and contributes to cancer cachexia. In collaboration with this group, we have shown that PTH also stimulates adipose tissue browning and causes cachexia in advanced CKD [29]. Furthermore, we have shown that adipocyte-specific PTH/PTHrP receptor knockout mice are resistant to adipose tissue browning and wasting even at high PTH levels induced by 5/6 nephrectomy.
These experimental data identified PTH as a driver of adipose tissue browning and wasting, but it remained unclear whether poorly controlled SHPT leads to increased energy expenditure and contributes to poor clinical outcomes. To address this, we analyzed data from the international DOPPS and found a strong linear correlation between baseline PTH levels and weight loss (a surrogate marker of wasting) over the subsequent 12 months [30]. This association remained significant after adjustment for numerous covariates and was robust regardless of recent hospitalization. Furthermore, the association between PTH levels and weight loss partially mediated the increased risk of mortality associated with elevated PTH levels. These findings suggest that this pathway may be a mediator between elevated PTH levels and mortality in patients undergoing dialysis.
Another possible mechanism linking SHPT to poor clinical outcomes may be through elevated circulating FGF23 levels. PTH is one of the main regulators of FGF23 secretion in osteocytes, and SHPT is crucial for maintaining high FGF23 levels [31]. We and others have reported that both calcimimetics and parathyroidectomy significantly decrease both circulating PTH and FGF23 levels in patients with SHPT undergoing dialysis [32,33], suggesting that the survival benefit associated with parathyroidectomy may be mediated by the reduction of both PTH and FGF23 levels. As described below, several experimental studies have demonstrated that FGF23 directly induces multiple-organ injuries. However, it remains to be determined whether FGF23 directly contributes to poor outcomes in patients with advanced CKD, and further studies are needed to confirm this possibility.
CURRENT MANAGEMENT OF SHPT
Until several years ago, vitamin D receptor activators (VDRAs) were the mainstay of treatment for SHPT. However, the clinical utility of VDRAs was limited because advanced parathyroid hyperplasia develops tolerance to these drugs due to decreased expression of the calcium-sensing receptor (CaSR) and vitamin D receptor in the parathyroid gland [34-36]. Calcimimetics, a relatively new option for the therapeutic control of SHPT, allosterically modulate the parathyroid CaSR and increase its sensitivity to extracellular calcium. Currently, calcimimetics are considered the first choice for SHPT in dialysis patients because they exert a strong inhibitory effect on PTH, leading to an effective reduction in parathyroid volume [37]. Two recent experimental studies evaluated the effects of calcimimetics on bone metabolism and showed that evocalcet [15] and etelcalcetide [36], which are novel calcimimetics, suppressed the progression of cortical porosity in severe SHPT. In the clinical setting, the Bone Histomorphometry Assessment for Dialysis Patients with Secondary Hyperparathyroidism of End-Stage Renal Disease (BONAFIDE) study showed that 6 to 12 months of treatment with cinacalcet reduced the elevated bone formation rate and improved bone histology [38]. Regarding the effect of calcimimetics on fracture risk, a subanalysis of the Evaluation of Cinacalcet Hydrochloride Therapy to Lower Cardiovascular Events (EVOLVE) trial, which found no significant effect of cinacalcet in the primary intention-to-treat analysis, showed a significant reduction in fracture risk in the cinacalcet group in the prespecified lag-censoring analysis [39]. The EVOLVE study also suggests that cinacalcet may improve survival. Although the primary analysis did not show a significant reduction in all-cause and cardiovascular mortality, the prespecified lag-censoring analysis showed a significant effect in patients treated with cinacalcet [40]. Taken together, these clinical trial data highlight the importance of controlling parathyroid function and bone turnover as a means of not only reducing fracture risk, but also improving survival.
POTENTIAL BENEFITS OF INTENSIVE PTH CONTROL
How tightly should we control PTH in patients with kidney failure? Since parathyroidectomy lowers PTH levels more drastically than calcimimetics, it stands to reason that comparing the long-term outcomes of these treatments could shed light on this issue. Therefore, we analyzed data from the nationwide registry of Japanese dialysis patients and compared patients who underwent parathyroidectomy to those who started treatment with cinacalcet after propensity matching at a 1:3 ratio [41]. Patients in the parathyroidectomy group had lower levels of intact PTH, calcium, and phosphorus than those in the cinacalcet group. During the 6-year follow-up period, parathyroidectomy was associated with a 22% lower risk of mortality compared with cinacalcet. As an exploratory analysis, we stratified patients undergoing parathyroidectomy into tertiles based on their postoperative intact PTH levels and performed propensity score matching for each tertile. The survival benefit of parathyroidectomy over cinacalcet was most evident in patients who exhibited a sustained reduction in postoperative PTH levels, supporting the concept of “the lower, the better” for the management of SHPT [42]. There may be concerns that excessive suppression of PTH following parathyroidectomy could lead to adynamic bone disease. However, we observed no difference in the incidence of hip fractures between the parathyroidectomy and cinacalcet groups. This finding suggests that the prevailing view that low PTH levels should be avoided to preserve bone strength may need to be reevaluated.
The target range for intact PTH levels in dialysis patients varies in different regions of the world. The Japanese Society for Dialysis Therapy guideline suggests maintaining intact PTH levels in the range of 60 to 240 pg/mL [43], whereas the Kidney Disease: Improving Global Outcomes (KDIGO) guideline suggests two to nine times the upper normal limit of intact PTH (approximately 130 to 585 pg/mL) [44]. Japanese hemodialysis patients show better survival outcomes than those in other regions [45], suggesting that more intensive PTH control may lead to better clinical outcomes. Further clinical studies are needed to determine the optimal PTH target in dialysis patients.
POTENTIAL OFF-TARGET EFFECTS OF FGF23 IN ADVANCED CKD
Effects of FGF23 on the parathyroid glands
FGF23 acts on the parathyroid glands and directly inhibits PTH secretion [46]. However, most patients with advanced CKD have high PTH levels despite markedly high FGF23 levels. To determine the mechanism underlying parathyroid resistance, we examined hyperplastic parathyroid tissue from patients undergoing parathyroidectomy [47]. We found that the expression of Klotho and its coreceptor FGF receptor 1 (FGFR1) significantly decreased, especially in glands with nodular hyperplasia (an advanced form of parathyroid hyperplasia). Other experimental studies using CKD models have also shown that FGF23 fails to suppress PTH secretion from parathyroid glands, presumably due to decreased expression of the Klotho-FGFR complex [48,49]. Of note, a recent experimental study using parathyroid-specific knockout mouse models demonstrated the importance of Klotho-CaSR interactions in PTH synthesis and parathyroid hyperplasia [50]. In that study, parathyroid-specific deletion of Klotho alone did not suppress an inhibitory effect of FGF23 on PTH secretion, but dual deletion of Klotho and CaSR led to increased PTH secretion and accelerated parathyroid hyperplasia compared to CaSR deletion alone. These findings suggest that in advanced CKD, decreased expression of Klotho and FGFR1, as well as CaSR, may induce parathyroid resistance to FGF23 and thus contribute to parathyroid hypersecretion and SHPT progression [1].
Potential effects of FGF23 on bone
Another potential target for FGF23 is bone, as the Klotho-FGFR1 complex is expressed at low levels in osteoblasts and osteocytes [51]. Although the role of Klotho in bone cells is not fully understood, recent experimental studies suggest that FGF23 acts directly on osteocytes via the Klotho-FGFR1 complex, thereby increasing the production of FGF23 itself and inhibiting bone formation [52,53]. Since Klotho expression in bone cells decreases in advanced CKD, it might induce skeletal resistance to FGF23. Further studies are needed to determine whether skeletal resistance to FGF23 can be a therapeutic target for renal osteodystrophy [54].
Off-target effects of FGF23 on the myocardium
As described above, FGF23 is unlikely to exert Klotho-dependent effects on classical target organs such as parathyroid glands and bone in advanced CKD due to the decreased expression of Klotho. In this setting, FGF23 might exert Klotho-independent effects on non-classical target organs [55]. Several experimental studies suggest that FGF23 has several pathogenic off-target effects, the most extensively studied of which is the possibility that FGF23 acts on cardiomyocytes via FGFR4 to induce left ventricular hypertrophy [56,57]. This finding lends biological plausibility to the observational evidence showing a strong association between high FGF23 levels and mortality [58-62].
However, there are several caveats to the hypothesis that elevated FGF23 levels are associated with mortality. First, the strength of the association between FGF23 and mortality is not consistent across the spectrum of CKD. In patients with predialysis CKD, the association between FGF23 and mortality would be expected to increase as kidney function declines [58]. However, it is progressively attenuated after the initiation of dialysis and almost disappears during long-term dialysis, despite remarkably high levels of FGF23 [59-61]. It then reappears after kidney transplantation despite a striking decrease in FGF23 [62]. Therefore, the changing pattern of FGF23-associated mortality does not parallel the trajectory of circulating FGF23 levels, which suggests that the relationship cannot be explained by a simple exposure-response mechanism. Second, cardiac hypertrophy has not been reported in disorders involving FGF23 excess, such as X-linked hypophosphatemia [63,64]. Another hypothesis is that the effects of FGF23 on the myocardium are enhanced in advanced CKD. However, this possibility seems unlikely because, as noted above, the association between FGF23 and mortality is not strengthened in dialysis patients. Finally, it should be noted that injured cardiomyocytes have the potential to produce FGF23. Experimental studies in animal models of myocardial infarction and cardiac hypertrophy showed a striking increase in FGF23 expression in cardiomyocytes [65,66]. An observational study showed a remarkable increase in circulating FGF23 levels in patients with cardiogenic shock [67]. In light of these findings, it is more plausible to suggest that cardiac injury induces an elevation in FGF23 levels rather than being a consequence of it. Although observational studies have reported an association between FGF23 and mortality, it remains unclear whether FGF23 has a direct cardiac effect. Further studies are needed to confirm this possibility [68].
The off-target effects of FGF23 are not limited to its potential to induce cardiac hypertrophy; other effects include the induction of inflammation [69], immune dysfunction [70], and anemia [71]. These toxic effects have been demonstrated in experimental data, but the clinical implications and relevance of these off-target effects are even less well understood.
CONCLUSIONS
Elevated levels of PTH and FGF23 play a pivotal role in the pathogenesis of CKD-MBD and are associated with several adverse outcomes, including fractures, cardiovascular disease and mortality in patients with advanced CKD (Fig. 1). Emerging evidence suggests that potential mechanisms linking SHPT and mortality may be partially explained by PTH-induced wasting. PTH-lowering therapy may improve mortality by attenuating PTH-induced wasting and other adverse effects, and our recent comparison of parathyroidectomy and cinacalcet treatment suggests the potential benefit of intensive PTH control. The target range for intact PTH levels remains a matter of debate, and further research is needed to determine the optimal PTH target in dialysis patients. Observational studies have also shown an association between elevated FGF23 and mortality, but it is not well understood whether FGF23 directly induces multiple-organ injury. Further studies are needed to determine whether intensive SHPT control and FGF23-targeted therapy improve clinical outcomes in CKD patients.
 XML Download
XML Download