

 PDF
PDF Citation
Citation Print
Print



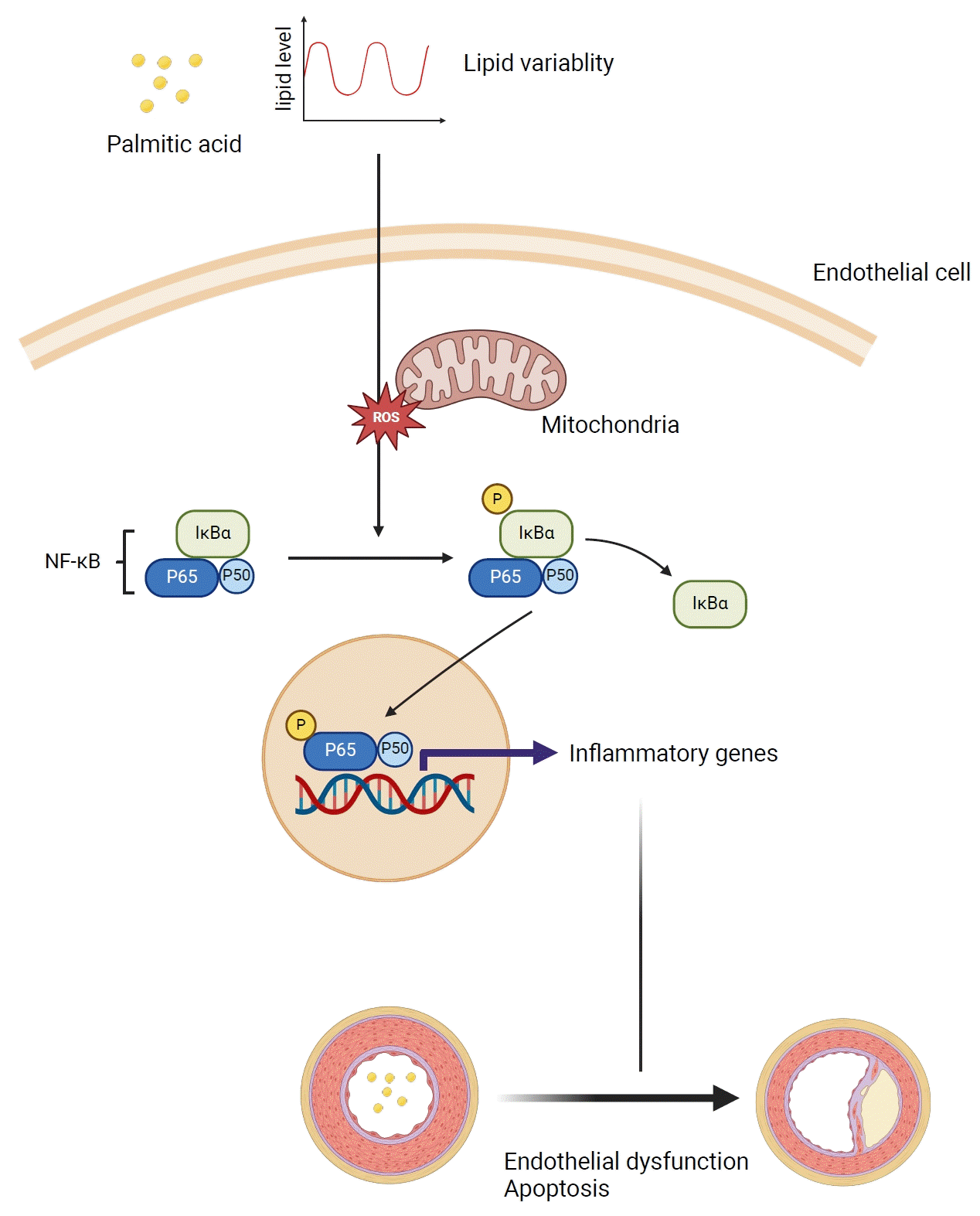
INTRODUCTION
Endothelial dysfunction, which is often caused by disruptions in lipid homeostasis, is a major initiator of atherosclerotic diseases. Endothelial cells (ECs), which form the inner lining of vascular surfaces, maintain vascular tone, facilitate blood flow, and regulate permeability [1]. Additionally, they play a vital role in regulating inflammation, hemostasis, thrombosis, fibrinolysis, and angiogenesis [2]. Central to EC functionality is the synthesis of diverse molecules that balance vasoconstriction (e.g., angiotensin [Ang] II and endothelin [ET]-1) and vasodilation (e.g., nitric oxide [NO] and bradykinin), creating a delicate equilibrium essential for circulatory system maintenance. This equilibrium safeguards against disturbances that may otherwise trigger the progression of vascular diseases [3]. Perturbations introduced by stimuli such as lipids, glucose, or blood pressure can induce EC dysfunction, which is characterized by diminished NO production and imbalanced ratios of endothelial vasodilators and vasoconstrictors. These deviations have emerged as precursors of cardiovascular risk and signal the onset of atherosclerosis [4]. Among the many stimuli, the disruption of lipid homeostasis is notable for being a precursor to endothelial dysfunction. Lipids are the main contributors to this cascade. Elevations in low-density lipoprotein cholesterol (LDL-C) levels and the cumulative extent of exposure are well-established cardiovascular risk factors [5-8]. Several studies have suggested an association between hypertriglyceridemia and cardiovascular risks [9-11]. Furthermore, elevated levels of serum-free fatty acids (FAs) have been implicated in an increased risk of cardiovascular diseases [12,13]. Notably, saturated FAs have been implicated in the development of atherosclerosis, a connection supported by mechanisms involving inflammation, oxidative stress, and impaired NO production in ECs [2]. Previous studies by our group and others have demonstrated that lipid variability is associated with the risk of cardiovascular diseases [14-16]; however, the precise mechanisms underlying the increased risk of atherosclerotic diseases resulting from fluctuations in lipid levels remains poorly understood. To address this, we explored the mechanism by which lipid variability leads to endothelial dysfunction using human EC lines and palmitic acids (PAs).
METHODS
Cell culture
Human aortic endothelial cells (HAECs; Lonza, Walkersville, MD, USA) and human umbilical vein endothelial cells (HUVECs) were used at passage 4–9 for the experiments and cultured in endothelial cell growth medium (EGM)-2 BulletKit (Lonza) supplemented with penicillin-streptomycin. The cells were incubated at 37°C in a humidified atmosphere containing 5% CO2. To analyze the molecular changes occurring under various PA treatment conditions, ECs were either treated with 0, 50, and 100 µM PA, or alternated between 0 and 100 µM PA every 8 hours (variability group) over 48 hours. The ECs in the lipid variability group were exposed to 100 μM of PA in the last 8 hours of culture before every transcriptomic, proteomic, and functional analysis to compare with continuous 100 µM PA group at the same condition.
Preparation of FA-bovine serum albumin complex
A 100 mM PA (P5585, Sigma-Aldrich, St. Louis, MO, USA) stock solution was prepared with 0.1 N NaOH at 70°C. The stock solution was conjugated to 10% FA-free bovine serum albumin (BSA; A7030, Sigma-Aldrich) in a heating block for 5 minutes at 60°C for complex formation. This intermediate stock was filter sterilized (0.22 μm) and the required amount was added to the medium for final concentrations.
Real-time quantitative polymerase chain reaction
The cells were treated with TRIzol reagent (Invitrogen, Carlsbad, CA, USA) to extract total RNA. In the reverse transcription reaction, 1 μg of total RNA was used with the PrimeScript 1st strand cDNA synthesis kit (Takara Bio Inc., Tokyo, Japan) following the manufacturer’s instructions. All quantitative polymerase chain reaction (qPCR) reactions were conducted using Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA). Supplemental Table S1 provides the details of the PCR primer sequences.
Western blot and immunocytochemistry
Western blot analysis was performed as described previously [17]. Total protein lysates were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto Immobilon polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). After blocking, the membranes were incubated with primary antibodies, and subsequently with horseradish peroxidase-conjugated secondary antibodies. The following primary antibodies employed were used: anti-phospho-nuclear factor κB (NF-κB) p65 (p-p65, #3031), anti-NF-κB p65 (p65, #3034), anti-phospho-IkappaB-alpha (p-IκBα, #9241), anti-IκBα (#9242), anti-intercellular adhesion molecule (ICAM, sc-7891), anti-vascular cell adhesion molecule (VCAM, sc8304), anti-E-selectin (sc-14011, Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-phospho-endothelial nitric oxide synthase (eNos, #9571), anti-B cell CLL/lymphoma 2 (BCL2, #3498), anti-BCL-2-associated X protein (BAX, #2774, Cell Signaling Technology Inc., Danvers, MA, USA), anti-eNos (610299, BD Bioscience, San Jose, CA, USA), anti-inducible nitric oxide synthase (iNOS, 1:1,000, N32030, Transduction Laboratories, Lexington, KY, USA), or anti-β-actin (1:5,000, A5441, Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Proteins were detected using an enhanced chemiluminescence kit (34577, Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. Visualization was performed using the PXi4 Multi-Application Gel Imaging System (Syngene, Cambridge, UK), and densitometry analysis was performed using ImageJ (National Institutes of Health, Bethesda, MD, USA), and GraphPad Prism 8.0 (San Diego, CA, USA). For immunofluorescence staining, cells were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS), permeabilized with 0.5% Triton X-100. Cells were then incubated with the anti-NF-κB p65 (p65, #8242, Cell Signaling Technology Inc.) overnight at 4°C. Cell nuclei were counterstained with 4´,6-diamidino-2-phenylindole (DAPI). Microscopy analysis was performed using an Olympus IX70 inverted fluorescence microscope (Olympus, Tokyo, Japan).
Secretory cytokines
Concentrations of secretory cytokines (interleukin [IL] 6 and monocyte chemoattractant protein [MCP] 1) were measured via Human Luminex Discovery Assay following the manufacturer’s instructions (#LXSAHM-08, R&D Systems, Minneapolis, MN, USA) using HUVEC supernatants.
Reactive oxygen species measurement
To quantify intracellular reactive oxygen species (ROS) levels, HAECs cultured under various conditions of PA exposure were cultured to 70% to 80% of confluency. The cells were then washed with PBS and incubated with 10 μM 2´,7´-dichlorofluorescin diacetate (DCFH-DA, D6883, Sigma-Aldrich) in serum-free medium for 30 minutes to 1 hour at 37°C in a humidified incubator at 5% CO2. After the incubation period, the cells were washed with PBS to remove any extracellular DCFH-DA and subsequently exposed to the experimental conditions. Fluorescence intensity was measured using a fluorescence microplate reader at excitation and emission wavelengths of 488 and 525 nm, respectively. Fluorescence readings were taken at specified time points, and the data were normalized to the control wells to determine the relative ROS levels.
Oxygen consumption rate measurement
Mitochondrial function in HAECs was measured using the mitochondrial stress test (Seahorse Bioscience, North Billerica, MA, USA) as described previously [17]. Briefly, cells were seeded at a density of 4×104 cells/well in an XF24 flux analyzer plate (Seahorse Biosciences). Mitochondrial inhibitors were added at the following concentrations: 1.0 μM oligomycin, 0.5 mM trifluoromethoxy carbonylcyanide phenylhydrazone, and 0.5 mM rotenone and antimycin A (Seahorse Bioscience).
Wound healing assay
HAECs were serum-starved for 6 hours under various PA exposure conditions to synchronize and minimize proliferation. A sterile 200-μL pipette tip was used to create a linear scratch (wound) across the cell monolayer. Detached cells and debris were gently washed away with PBS, and fresh EGM was added to the wells. Cells were incubated at 37°C in a 5% CO2 atmosphere. Photomicrographs were captured at 0 and 48 hours postscratching using an inverted microscope to monitor wound closure. The distance between the edges of the scratch was measured and quantified using image analysis software to assess the cell migration rates.
Cell adhesion assay
The HAECs were cultured in 12-well plates under various PA exposure conditions. The medium was replaced with serum-free EGM for 6 hours to synchronize the cells. THP1 monocytes (1×106 cells/mL) were pre-stimulated with calcein-acetoxymethyl (AM) (2.5 μg/mL, 17783, Sigma-Aldrich) for 30 minutes. Subsequently, calcein-AM-labeled monocytes were added to the HAEC monolayer and co-incubated for 1 hour at 37°C in a 5% CO2 atmosphere. Non-adhered monocytes were gently washed with PBS, and the adhered cells were quantified using a fluorescence microplate reader (Synergy MX, BioTek, Winooski, VT, USA) with excitation and emission wavelengths set at 495/515 nm. The data were normalized to control wells containing only ECs to calculate relative adhesion rates.
Cell viability assay
HAECs were seeded onto plates (2×104 cells/well in a 96-well plate) and cultured under various PA exposure conditions for 48 hours. Medium containing BSA was used as a blank. After PA exposure, 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT; M2128, Sigma-Aldrich) reagent (20 μL, 5 mg/mL) was added to each well and cells were incubated at 37°C for 4 hours. The absorbance was measured at 570 nm using a fluorescence microplate reader. Cell viability was calculated as follows: cell viability (%)=absorbance of the experimental group/control group×100.
RESULTS
Lipid variability induces inflammation in human ECs
Endothelial inflammation is an early event of endothelial dysfunction. This led us to hypothesize that lipid level fluctuations may increase inflammatory signals in ECs. To test this hypothesis, we exposed HAECs and HUVECs to PA at concentrations of 0, 50, and 100 µM, or alternated between 0 and 100 µM every 8 hours (variability group) over a 48-hour period (Fig. 1A). PA treatment consistently upregulated inflammatory genes, such as Il6, Mcp1, Icam, Vcam, E-selectin, and iNos, while downregulating eNos in HAECs (Fig. 1B). Notably, Mcp1, Vcam, and Eselectin showed even greater upregulation in the variability group compared to the 100 µM PA-treated HAECs. Similar upregulation in inflammatory genes (Il6, Mcp1, Icam, Vcam, E-selectin) were observed in PA-treated HUVECs (Supplemental Fig. S1A). Corresponding to the transcriptomic changes, the protein levels of ICAM, VCAM, E-selectin, and iNOS were significantly increased in the variability group, surpassing the levels observed in the 50 µM PA-treated group and similar to the 100 µM PA-treated group (Fig. 1C, D). Similarly, levels of secreted inflammatory cytokines (IL6, MCP1) were substantially elevated in the variability group compared to the control and 50 µM PA-treated group (Fig. 1E). NF-κB is a transcription factor that mediates inflammatory reactions in various pathophysiological contexts [18]. To further explore the role of NF-κB signaling in the inflammatory response of ECs under different lipid exposure conditions, we conducted Western blot analysis and immunofluorescence staining. We observed an increase in phosphorylation of p65 and IκBα (p-p65/p65 and p-IκBα/IκBα) (Fig. 1F), along with the translocation of NF-κB p65 from the cytoplasm to the nucleus (Fig. 1G) in ECs exposed to lipid variability conditions. These findings suggest that lipid variability activates the NF-κB signaling pathway, leading to human endothelial inflammation.
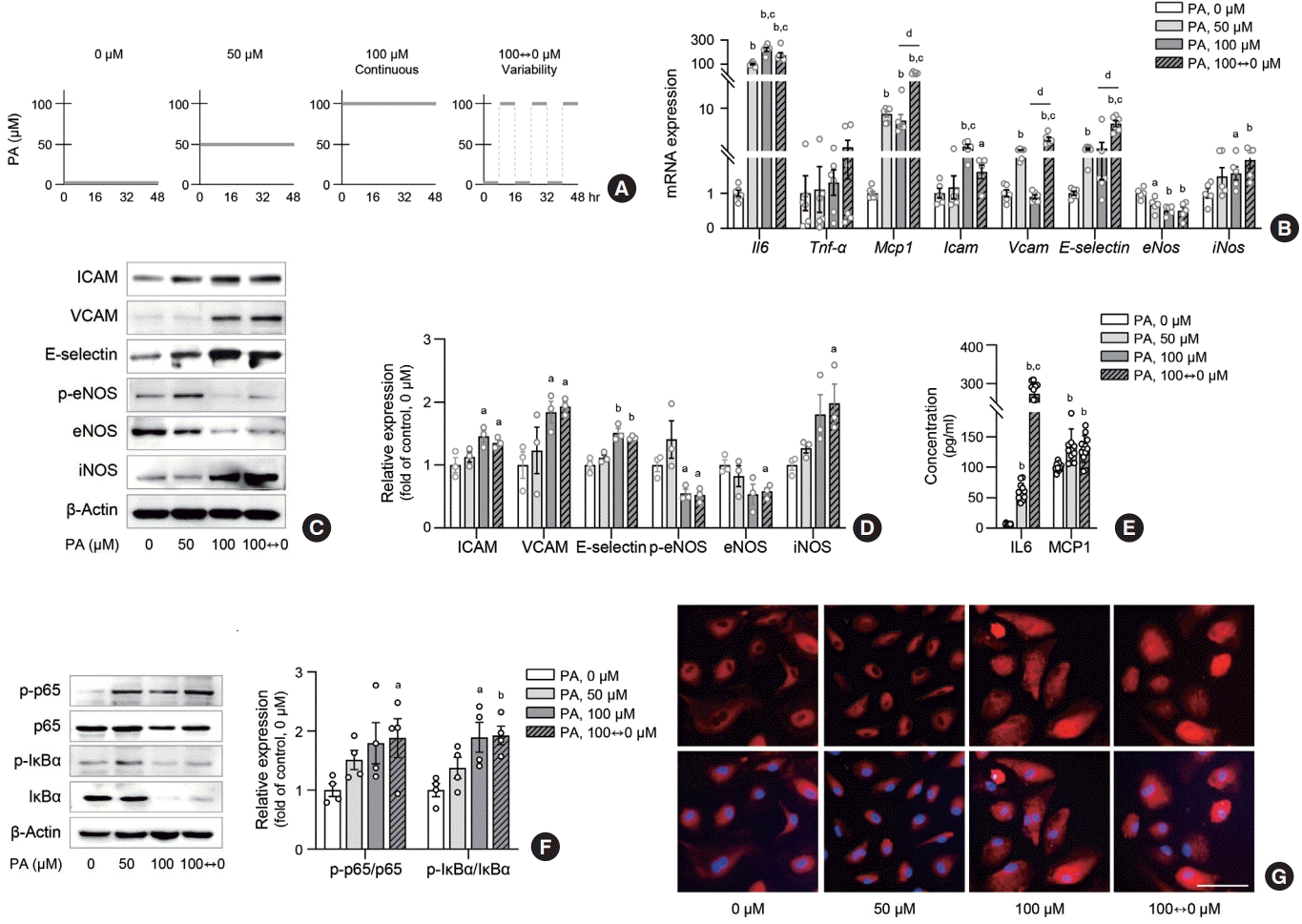
Fig. 1.
Lipid variability increases inflammation in human endothelial cells. Human umbilical vein endothelial cell (HUVEC) and human aortic endothelial cell (HAEC) were exposed to 10% bovine serum albumin as a vehicle control or different concentrations of palmitic acid (PA) for 48 hours: continuous 0, 50, and 100 μM, or alternating 0 and 100 μM every 8 hours (A). The expression of genes (B) and proteins (C, D) associated with inflammation and adhesion molecules were determined in HAEC after PA treatment. Interleukin 6 (IL6) and monocyte chemoattractant protein-1 (MCP1) concentrations were measured by enzyme-linked immunosorbent assay (ELISA) in cell supernatants from HUVEC (E). The nuclear factor κB (NF-κB) subunits were determined in HAEC after PA treatment (F). Nuclear translocation of NF-κB p65 (red fluorescence) and 4′,6-diamidino-2-phenylindole (DAPI)-stained nuclei were detected by fluorescence staining in HAEC (G). Scale bar=100 μm. Data are presented as the mean±standard error of the mean of three to five independent experiments. Tnf-α, tumor necrosis factor-α; ICAM, intercellular adhesion molecule; VCAM, vascular cell adhesion molecule; eNOS, endothelial nitric oxide synthase; iNOS, inducible nitric oxide synthase; IκBα, IkappaB-alpha. aP<0.05 and bP<0.01 compared to untreated control; cP<0.01 compared to 50 μM; dP<0.01 compared to continuous 100 μM PA.
![]()
Lipid variability increases oxidative stress in human ECs
Because oxidative stress commonly underlies inflammation in various pathological contexts, we postulated that it could mediate endothelial inflammation triggered by lipid variability. To confirm whether oxidative stress increased in the lipid variability group, we measured the intracellular levels of ROS in human ECs using the DCFH-DA assay. The fluorescence intensity in the lipid variability group was higher than that of the control or 50 µM PA-treated group, and was comparable to the intensity observed in the 100 µM PA-treated group (Fig. 2A, B). Additionally, the transcriptomic level of heme oxygenase (Ho) 1 was elevated by variable lipid levels to a degree comparable to that of the 100 µM PA-treated group (Fig. 2C, Supplemental Fig. S1B).
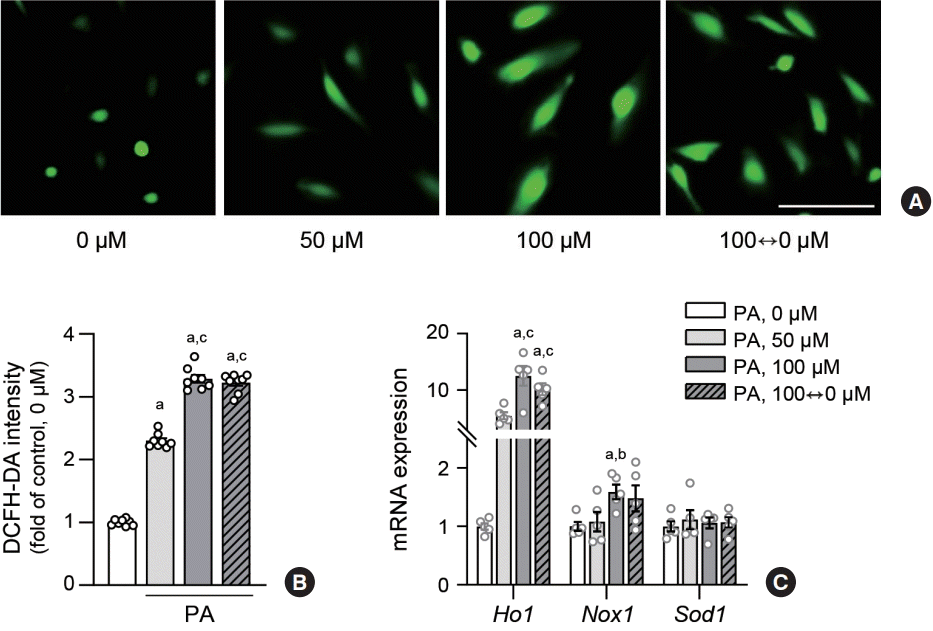
Fig. 2.
Lipid variability increases oxidative stress in human endothelial cells. Human aortic endothelial cells (HAECs) were treated with palmitic acid (PA) for 48 hours. Reactive oxygen species production levels were visualized (A) using fluorescence microscopy and quantified (B) after 1 hour incubation with 10 μM 2’,7’-dichlorofluorescin diacetate (DCFH-DA) probe. Scale bar=100 μm. The expression level of mRNA related nitric oxide synthase were measured using real-time polymerase chain reaction in HAEC (C). Data are presented as the mean±standard error of the mean of five independent experiments. aP<0.01 compared to untreated control; bP<0.05 and cP<0.01 compared to 50 μM PA.
![]()
Lipid variability leads to mitochondrial dysfunction in human ECs
Increased ROS levels are often preceded by mitochondrial dysfunction in pathological conditions. To investigate the effects of lipid variability on mitochondrial metabolism in ECs, we examined the cellular oxygen consumption rate (OCR) of HAECs under various PA treatments (Fig. 3A). Cells incubated with 100 µM PA and alternating 0 and 100 µM PA showed a lower OCR compared to the control and 50 µM PA-treated groups (Fig. 3B). Mitochondrial respiration, including basal and maximal respiration, adenosine triphosphate synthesis, and spare respiratory capacity, was also reduced in the lipid variability group (Fig. 3C). These findings suggest that lipid variability induces mitochondrial dysfunction, leading to ROS accumulation in ECs.
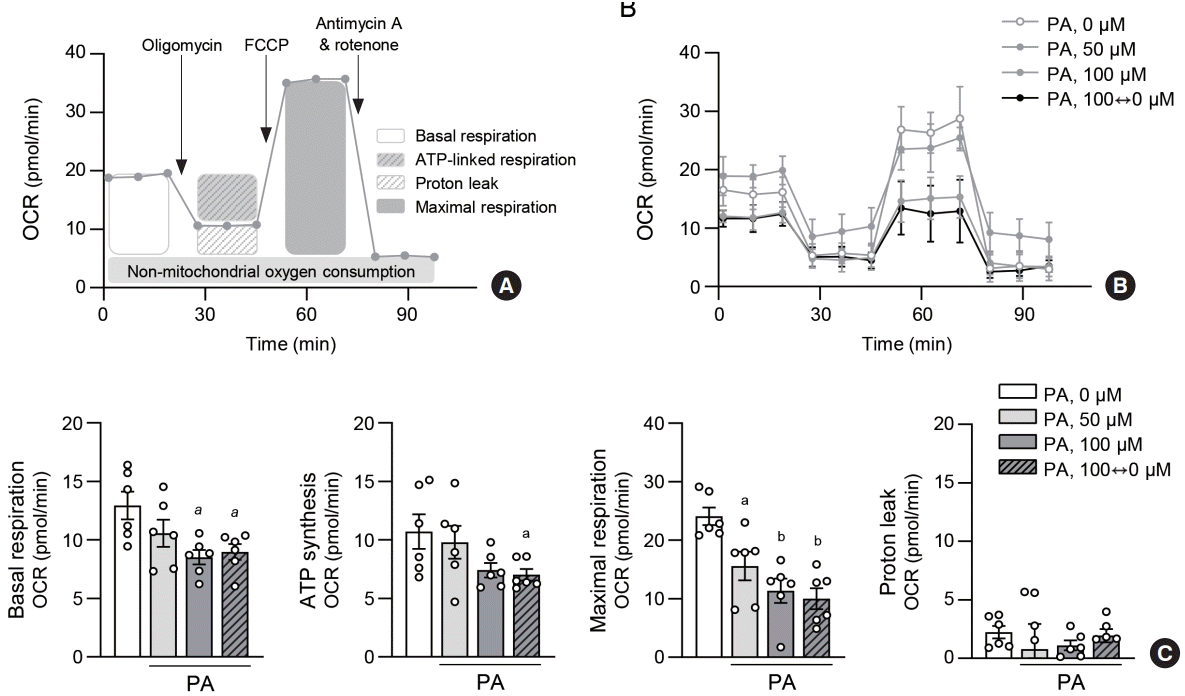
Fig. 3.
Lipid variability leads to mitochondrial dysfunction in human endothelial cells. For real-time measurements of the cellular oxygen consumption rate (OCR), human aortic endothelial cell (HAEC) were plated at 400,000 cells/well in XF24 cell multi-well plate. Cells were incubated with different concentrations of palmitic acid (PA) for 48 hours. Protocol used in data collection and calculations for evaluating OCR values (A). Representative mitochondrial respiration after PA exposure (B). Quantitative analysis of basal respiration, adenosine triphosphate (ATP)-linked respiration, maximal respiration and proton leak (C). Data are presented as the mean±standard error of the mean of five independent experiments. FCCP, carbonyl cyanide-4 (trifluoromethoxy) phenylhydrazone. aP<0.05 and bP<0.01 compared to untreated control.
![]()
Lipid variability induces human endothelial dysfunction
To evaluate whether increased inflammation translates to functional changes in ECs, we conducted wound healing assays on HAECs subjected to varying PA concentrations. Cell migration area was reduced in the lipid variability group compared to the control or 50 µM PA-treated group (Fig. 4A, B). This finding was corroborated by monocyte adhesion assays, where fluorescence-labeled THP1 cells exhibited increased adhesion to HAECs in the lipid variability group, indicating stimulation of monocyte adhesion due to lipid level fluctuations (Fig. 4C, D). These data indicated that lipid variability induces functional abnormalities in human ECs.
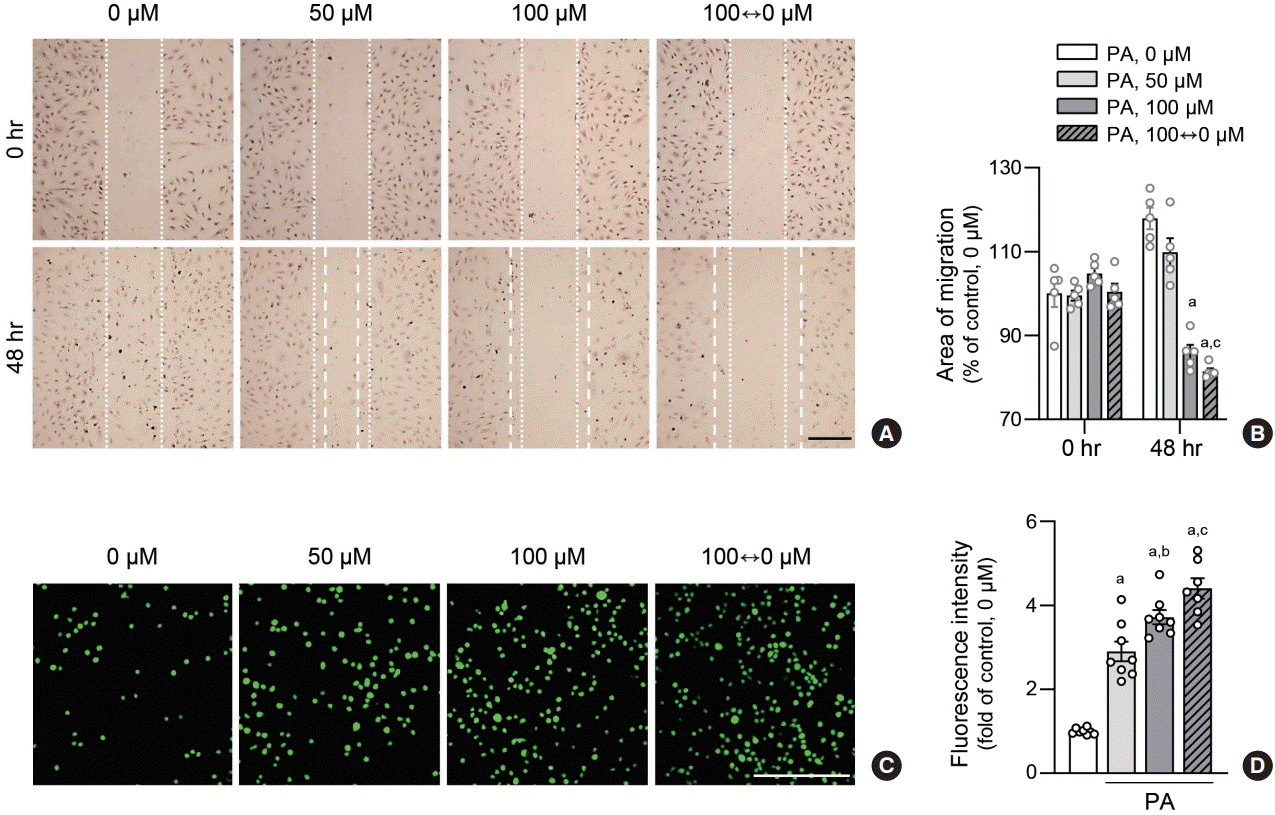
Fig. 4.
Lipid variability induces human endothelial dysfunction. Human aortic endothelial cells (HAECs) were exposed to various concentrations of palmitic acid (PA) for 48 hours. To analyze migration of endothelial cells treated with PA, wound healing assays were performed at indicated doses in HAEC. The dotted lines show the initial area without cells (A, B). Scale bar=200 μm. Data are presented as the mean± standard error of the mean of five independent experiments. HAEC were co-cultured with calcein-acetoxymethyl (AM)-labeled THP1 cells for 2 hours. THP1 monocytes that adhered to HAEC were visualized using a fluorescence microscope and counted (C, D). Scale bar=200 μm. Data are presented as the mean±standard error of the mean of three or five independent experiments. aP<0.01 compared to untreated control; bP<0.05 and cP<0.01 compared to 50 μM PA.
![]()
Lipid variability increases human endothelial apoptosis
We investigated whether lipid variability induces cell death in human ECs. MTT assays demonstrated a decrease in viable cell counts owing to variable lipid levels (Fig. 5A, Supplemental Fig. S1C). The expression of the apoptotic signal BAX was upregulated, whereas that of BCL2 was downregulated in response to lipid variability (Fig. 5B). Overall, lipid variability reduced cell viability by triggering apoptotic signals in human ECs.
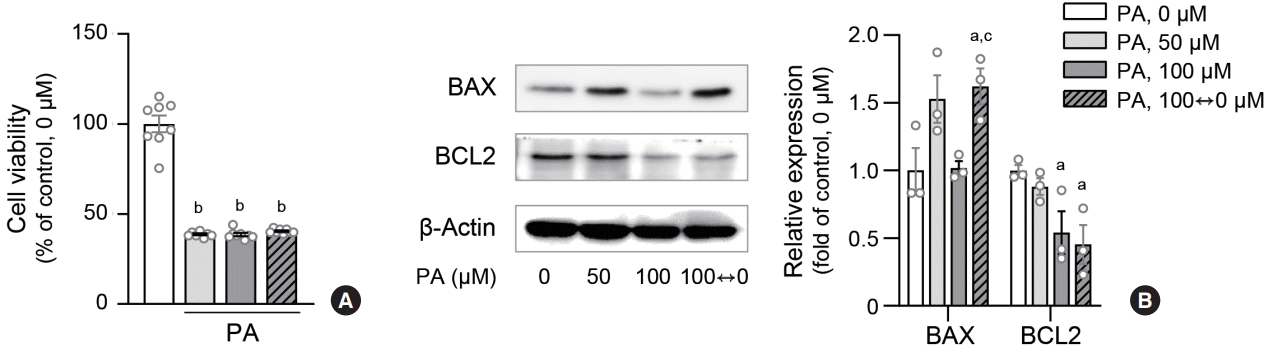
Fig. 5.
Lipid variability induces apoptosis in human endothelial cells. Cell viability of human aortic endothelial cells (HAECs) after palmitic acid (PA) exposure was measured using 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assay (A). Protein expression levels of BCL-2-associated X protein (BAX) and B cell CLL/lymphoma 2 (BCL2) were estimated by immunoblotting (B). Data are presented as the mean±standard error of the mean of three or five independent experiments. aP<0.05 and bP<0.01 compared to untreated control; cP<0.05 compared to continuous 100 μM PA.
![]()
DISCUSSION
In this study, we demonstrated that lipid variability induces endothelial dysfunction by increasing oxidative stress and inflammatory signaling. By treating HAECs and HUVECs with various concentrations of PAs, we consistently demonstrated that variability in lipid levels triggers endothelial dysfunction.
Advancements in lipid-lowering agents have substantially reduced the incidence of cardiovascular diseases [19-23]. However, lipid variability has emerged as an important factor in cardiovascular disease. Despite accumulating clinical evidence [14-16,24], the precise mechanisms by which lipid variability leads to the development of atherosclerotic diseases remain elusive. To the best of our knowledge, this is the first study to delineate the mechanistic link between lipid variability and endothelial dysfunction.
Notably, the degree of alterations in inflammatory genes, endothelial dysfunction, and ROS were consistently amplified in the variability group compared to both the control and the 50 μM PA-treated groups. These changes were comparable to those observed in the 100 μM PA-treated group. Mcp1, Vcam, and E-selectin transcription level was even higher in the variability group compared with the 100 μM PA-treated group. This observation led us to speculate that fluctuations in lipid levels create a distinct milieu that intensifies ROS-driven inflammation and ultimately contributes to endothelial dysfunction.
This study had certain limitations that we were unable to address. Although we demonstrated that lipid variability induces ROS, inflammation, apoptosis, and functional impairment in ECs, we could not directly establish the links between ROS, inflammation, and endothelial dysfunction. The correlation between ROS levels, inflammation, and endothelial dysfunction has been extensively explored in previous studies [25]. In response to stimuli such as endotoxins and cytokines, ECs increase the NF-κB expression, which subsequently upregulates effectors such as VCAM-1, E-selectin, and ICAM [26-28]. This upregulation of effector proteins on the endothelial surface facilitates the recruitment of monocytes and T lymphocytes to the subendothelial region. Activated ECs, smooth muscle cells, monocytes/macrophages, and lymphocytes form a complex local milieu of cytokines, growth factors, and ROS within the arterial wall that perpetuates chronic inflammation and drives the progression of atherosclerotic lesions. In our study, the expression of MCP1, ICAM, VCAM, E-selectin, and eNOS increased in human ECs due to the fluctuation of lipid variability. ICAM, VCAM, E-selectin, and eNOS are part of the pro-inflammatory endothelial gene complex and play a critical role in endothelial dysfunction [25]. ICAM, VCAM, and E-selectin are cell adhesion molecules that participate in angiogenesis [29,30]. We speculated that this altered gene expression contributed to the outcomes observed in the wound healing assay. MCP1 recruits monocytes and macrophages to ECs [31]. The elevated secretion of MCP1 was consistent with observations made in the cell adhesion assay. eNOS catalyzes NO production from L-arginine, and its decreased expression is indicative of endothelial dysfunction. Collectively, our data provide information that establishes a link between lipid variability, inflammation, and endothelial dysfunction.
ROS, which include oxygen ions and peroxides, are highly reactive molecules that are often generated as byproducts of cellular metabolism [32]. Excessive ROS results in oxidative stress, which adversely affects cellular components including proteins, DNA, and lipids. Notably, ROS triggers redox-sensitive signaling pathways that stimulate the production of pro-inflammatory molecules, thus creating an inflammatory environment [25]. This cascade reduces NO levels and increases endothelial apoptosis [33,34]. Our data demonstrated increased ROS levels in ECs under various lipid conditions. We speculate that this could have provided a pro-inflammatory environment, leading to decreased eNOS expression and increased apoptosis in ECs. However, we admit that the ROS-independent mechanism may have contributed to lipid variability-mediated endothelial dysfunction.
In the present study, we focused solely on the effects of fluctuations in PA levels on ECs. Further investigation is necessary to confirm whether fluctuations of other types of lipid (e.g., LDL-C, high-density lipoprotein cholesterol, triglycerides, and apolipoproteins) exert similar effects on ECs. Furthermore, as our study was conducted in vitro, additional in vivo investigations are needed to corroborate the physiological relevance of our findings.
In conclusion, our study demonstrated that lipid variability induces endothelial dysfunction by intensifying inflammation and oxidative stress. We speculate that these data provide valuable translational insights into how fluctuations in lipid levels are associated with an increased risk of cardiovascular diseases. Future in vivo studies investigating the mechanistic links between various lipid types and endothelial dysfunction would improve the robustness of our findings.
 XML Download
XML Download