

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Blastoid plasma cell leukemia (PCL) is a highly uncommon and aggressive plasma cell neoplasm, characterized by the presence of plasma cells exhibiting blastoid morphology, indistinguishable from leukemic blasts. Historically, only a handful of blastoid PCL cases have been reported in the United States [1, 2]. In this case report, we present the inaugural patient diagnosed with blastoid PCL in Korea, aiming to augment the limited understanding of this rare disease. Approval for this study was obtained from the Institutional Review Board (IRB) of Severance Hospital (IRB No. 4-2023-0668), and informed consent from the patient was waived.
Go to : 
CASE REPORT
At the time of initial diagnosis (Table 1, specimen number 1), a 74-year-old male patient presented with back pain, and his MRI scan revealed multifocal lesions in the skull. Subsequent bone marrow aspirate analysis revealed a mixture of mature and slightly immature-looking plasma cells, constituting 80.8% of the total nucleated cells (Fig. 1). Bone marrow biopsy demonstrated diffuse aggregates of kappa-restricted plasma cells. The serum kappa/lambda (K/L) ratio was 100.28, and serum M protein was undetectable. Immunofixation of IgD and IgE revealed no monoclonal band. Fluorescence in situ hybridization testing indicated TP53 deletion and IGH/CCND1 rearrangement. Based on these findings, the patient was diagnosed with plasma cell myeloma (PCM).
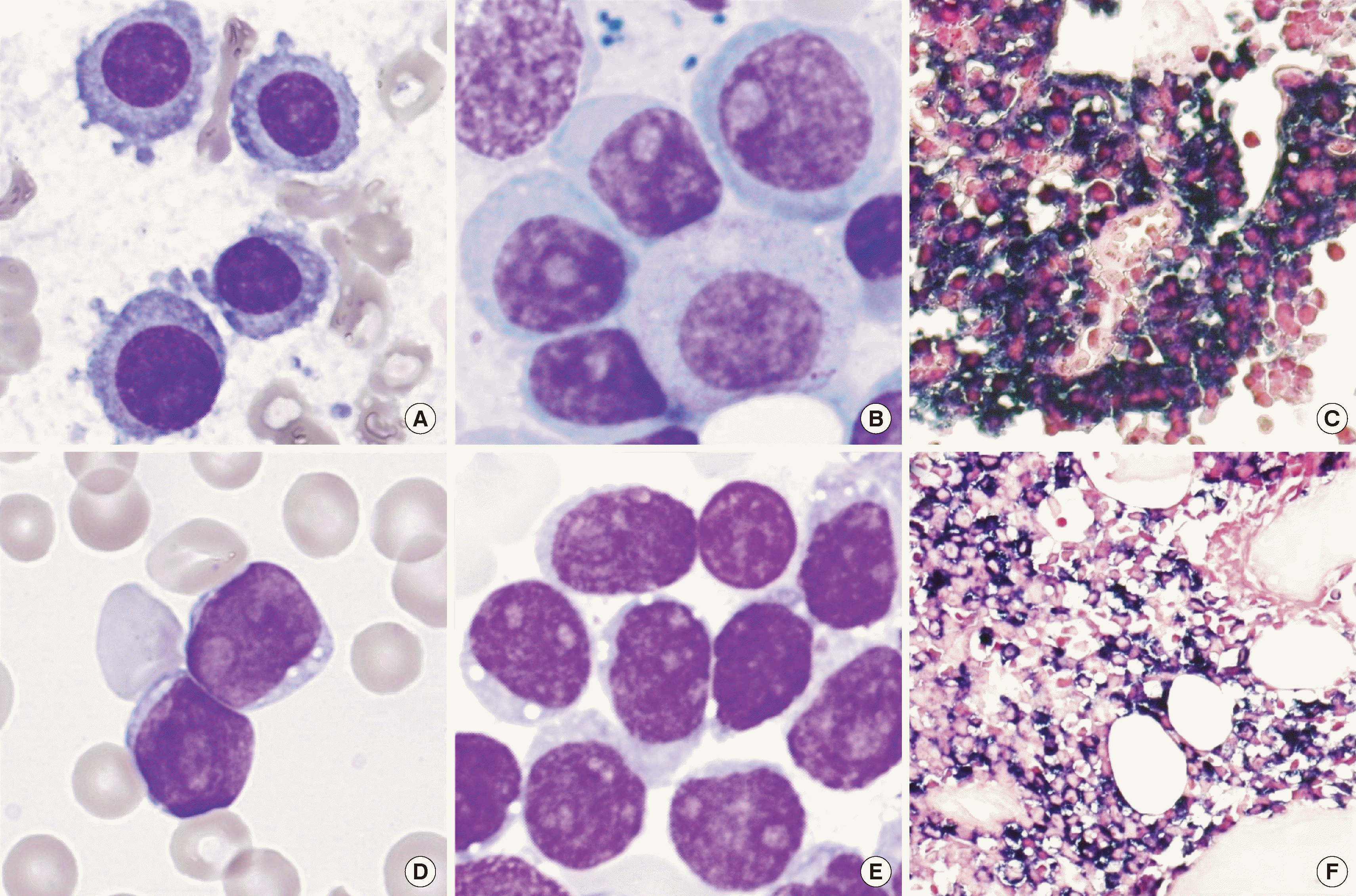 | Fig. 1Morphology of plasma cell myeloma (PCM) at initial diagnosis and secondary blastoid plasma cell leukemia (PCL). At the initial diagnosis of PCM, (A) mature plasma cells with coarse chromatin and abundant cytoplasm, and (B) slightly immature-looking plasma cells with moderately coarse chromatin, prominent nucleoli, and abundant cytoplasm were mixed in bone marrow aspiration (1000×, Wright-Giemsa stain). Additionally, bone marrow biopsy demonstrates (C) kappa light chain restriction (400×, in situ hybridization for kappa). In the state of PCL, (D) immature-looking plasma cells with fine chromatin, prominent nucleoli, and scant cytoplasm were observed in the peripheral blood smear, and (E) bone marrow aspiration (1,000×, Wright-Giemsa stain). Moreover, bone marrow biopsy shows (F) hypercellular marrow with blastoid plasma cells exhibiting kappa light chain restriction (400×, in situ hybridization for kappa).
|
Table 1
Laboratory, cytogenetic, and molecular characteristics of the patient
![]()
In the current case of PCL (Table 1, specimen number 4), despite treatment with lenalidomide, dexamethasone, and zoledronic acid, the patient exhibited refractory PCM. Peripheral blood smear and bone marrow aspirate analyses revealed a substantial number of immature cells characterized by fine chromatin, scant cytoplasm, and some nucleoli, constituting 40.0% and 84.3% of the respective cell populations. Furthermore, a complete blood count test indicated leukocytosis (white blood cells, 74.9×109/L), anemia (hemoglobin, 10.2 g/dL), and thrombocytopenia (platelets, 29×109/L). Flow cytometric analysis revealed a cell population exhibiting negative-to-intermediate CD45 expression and dim positivity for CD38 and CD138. Additional markers including CD34, CD117, myeloperoxidase, CD11c, CD33, CD14, HLA-DR, CD10, CD19, CD20, cytoplasmic CD22, CD3, CD4, CD8, CD7, CD56, CD36, CD41, and CD61 were negative. Bone marrow biopsy depicted a densely packed marrow with CD138-positive and kappa-restricted immature-looking cells. The serum K/L ratio was 211.78. Collectively, these findings confirmed the diagnosis of secondary PCL characterized by blastoid plasma cells.
The patient harbored a complex karyotype of 41–43,X,-Y,der(1)t(1;?)(q21;?),der(5)t(5;?)(q31;?),-8,t(11;14)(q13;q32),der(13)t(13;?)(q34;?),-14,-15,-15,+16,der(17)t(17;?)(p12;?),-20,-22,+1–4mar[cp19]/46,
XY[1], indicating genomic instability commonly associated with aggressive hematological malignancies. Subsequent next-generation sequencing (NGS) unveiled whole gene deletions encompassing TP53, RB1, TRAF3, DIS3, NFKBIA, and KMT2C, alongside a TP53 mutation (NM_000546.6:c.376-1G>C) with a variant allele frequency (VAF) of 93%. Additionally, mutations in TET2 (NM_001127208.3:c.2083dup, p.(Met695AsnfsTer17)), LRP1B (NM_018557.3:c.12947C>G, p.(Thr4316Ser)), and ARID5B (NM_
032199.3:c.3170C>T, p.(Ala1057Val)) were identified. The IGH/IGK clonality test using LymphoTrack (InVivoScribe Technologies, San Diego, CA, USA) revealed persisting rearrangements involving IGHV4-34/IGHJ4, IGKV1-5/IGKJ1, and IGKV2-29/IGKJ2 during disease progression from PCM to PCL.
Go to :
DISCUSSION
While blastoid PCL is indeed rare, its differential diagnosis from acute leukemia is feasible through various diagnostic tests such as immunohistochemistry or in situ hybridization, fiow cytometry, free light chain assay, and M-protein testing. Therefore, conducting comprehensive diagnostic evaluations is crucial. Particularly during the follow-up of PCM patients, the presence of immature-looking cells in the peripheral blood warrants consideration of both PCL and acute leukemia.
In this case, NGS indicated biallelic inactivation of TP53, demonstrated by both whole gene deletion and a TP53 mutation with a high VAF of 93%. Remarkably, the VAF of the TP53 mutation significantly increased during progression from PCM to PCL, rising from 8% to 93%, suggesting its potential role in PCL transformation (Table 1). Deletions and mutations affecting the TP53 gene are frequently observed in patients with PCM and PCL [3, 4], and these genetic alterations are associated with shorter median event-free survival and overall survival [4]. Additionally, it has been reported that the presence of del(17p) with biallelic TP53 inactivation in patients with PCM correlates with a poorer prognosis [5]. The patient succumbed to pneumonia, acute kidney injury, and septic shock on the 23rd day following PCL diagnosis.
Two previously reported blastoid PCL cases documented a complex karyotype with TP53 deletion, a genetic aberration also observed in this patient. However, unlike these cases, which exhibited blastoid plasma cells characterized by a moderate amount of cytoplasm or Auer-rod-like inclusions, our case displayed immature cells with scant cytoplasm. This observation suggests that blastoid PCL may manifest with diverse morphological features [1, 2].
In summary, we presented a rare case of blastoid PCL, marking the first domestic case report from Korea. Our findings underscore the importance of conducting comprehensive diagnostic evaluations, as blastoid PCL may be morphologically misidentified as other types of acute leukemia.
Go to :
 XML Download
XML Download