

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The accurate detection of genetic mutations is becoming increasingly important in diagnosing and managing acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL). Many mutations, such as breakpoint cluster region (BCR)::Abelson murine leukemia viral oncogene homolog 1 (ABL1) rearrangement, have been shown to represent distinct entities with different characteristics and prognoses. Consequently, various genetic assays from conventional karyotyping and reverse-transcriptase polymerase chain reaction (RT-PCR) to comprehensive next-generation sequencing (NGS) have been adopted in the field of hemato-oncology.
The genetic analysis of acute leukemia may yield false negative results because of various reasons. One thing to consider is that the tumor fraction in the sample affects the proportion of DNA containing somatic mutations. If the blast percentage is low in the bone marrow sample, DNA from cancer cells will be diluted by DNA from normal cells, thus increasing the probability of false negative results.
In this study, we present a case of B-cell ALL (B-ALL) where genetic mutations were not captured at initial diagnosis but were identified at relapse. The retrospective review revealed that some of the mutations were present even at the initial diagnosis but were hidden because of low tumor fraction.
CASE REPORT
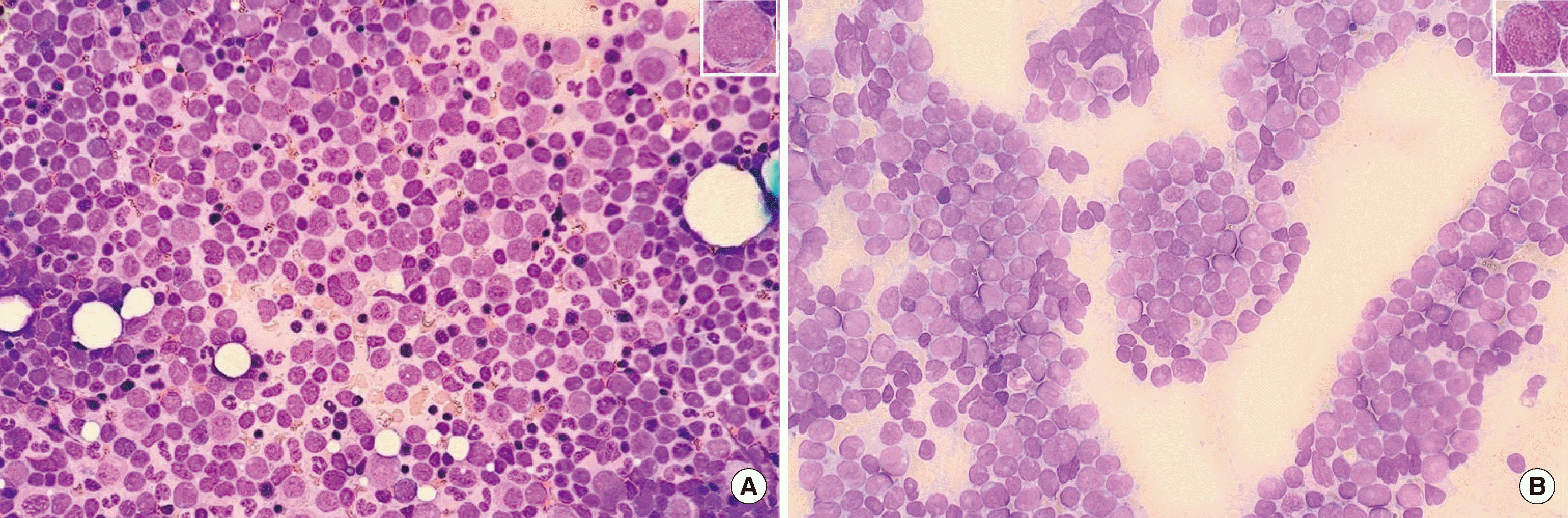
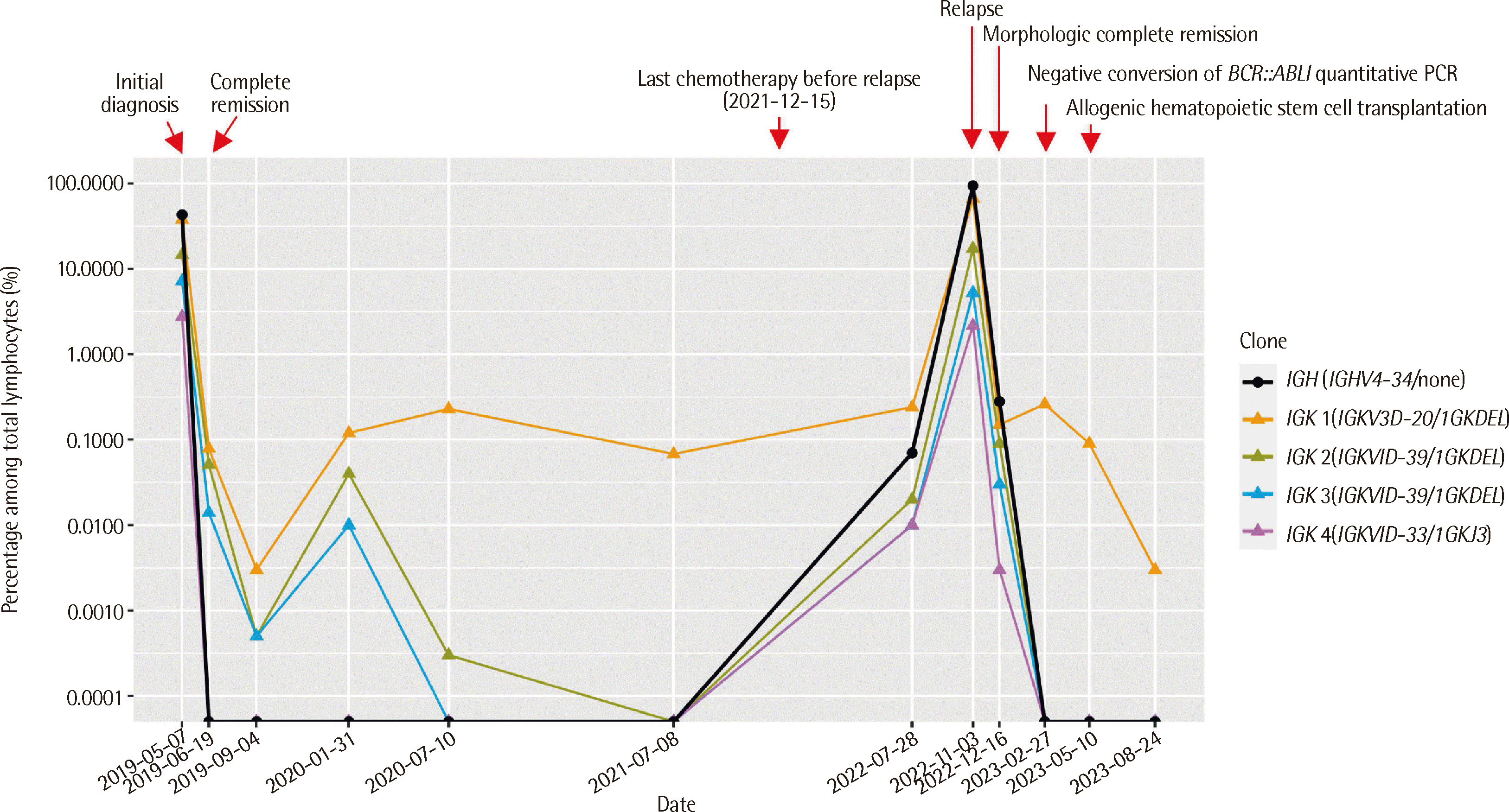
A 12-year-old female presented with fever and bone pain in May 2019. Whole body magnetic resonance imaging (MRI) and positron emission tomography/computed tomography showed multifocal bone marrow edema with fiuorodeoxyglucose uptake but without any definite mass lesion. Complete blood cell count revealed the following: white blood cells, 3.87×109/L; hemoglobin, 119 g/L; and platelets, 307×109/L. Blasts were not observed from peripheral blood smear; however, in bone marrow aspirate, blasts were 7.1% of total nucleated cells (Fig. 1A, Table 1). Although this was not a high proportion, the diagnosis of B-ALL was made because the fiow cytometric immunophenotyping of the blasts yielded positive results for immature B-cell markers (Table 2). The G-banding karyotyping and NGS targeting of 497 genes related to hematologic malignancy were performed using bone marrow aspirate, but no significant mutations were detected. She achieved complete remission in June 2019 and underwent consolidation chemotherapy until December 2021. However, immunoglobulin heavy chain (IGH)/kappa light chain (IGK) gene clonality assay still indicated measurable residual disease (MRD) (Fig. 2). IGH clone was negative near the last chemotherapy and increased to 0.07% in July 2022. IGK clone was positive for one clone, 0.068% near the last chemotherapy, and positive for all previously detected clones, with the largest being 0.24% in July 2022.
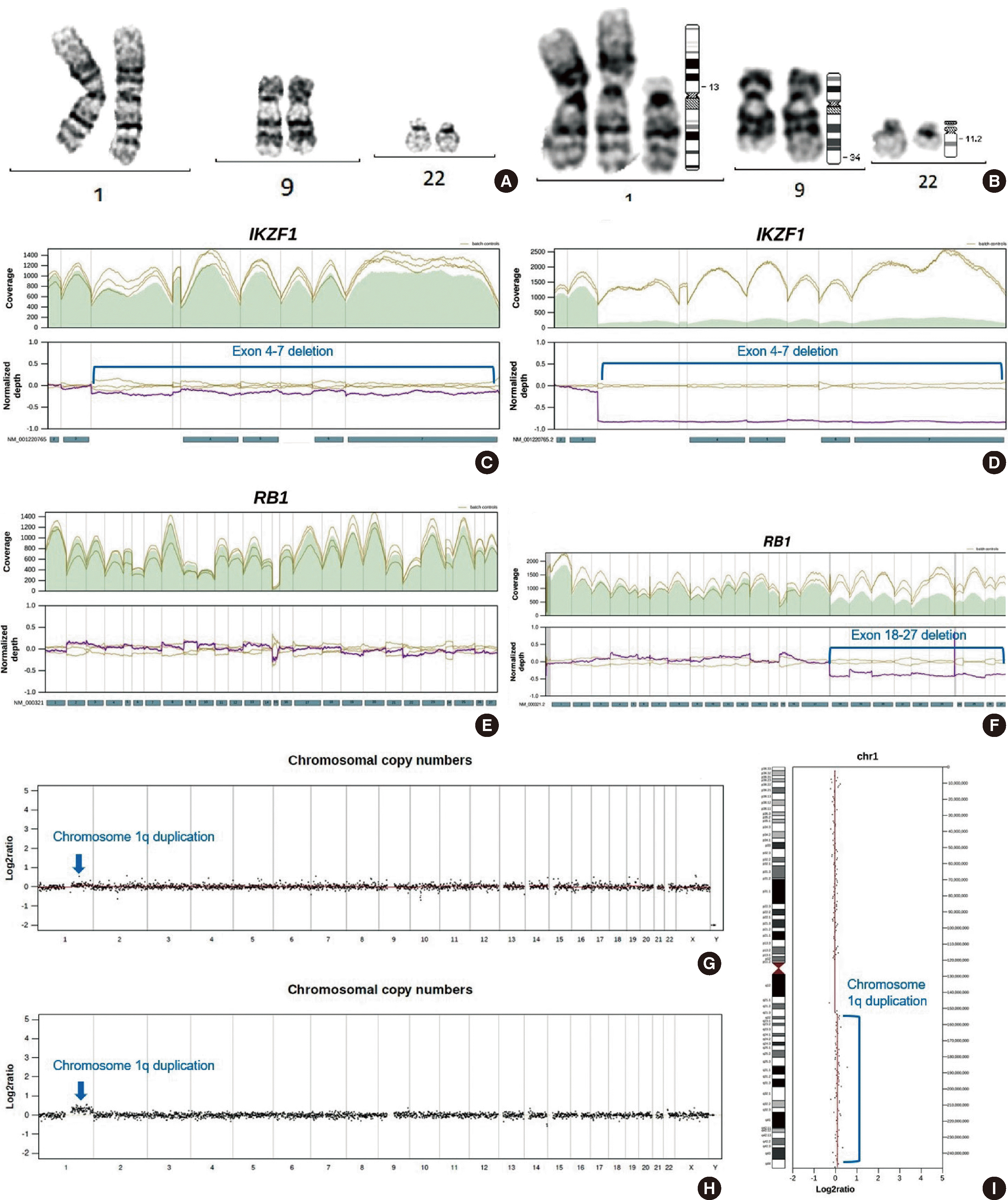
The patient revisited our hospital with chest wall pain in October 2022. Whole body MRI showed diffusely increased T2 signal intensity and increased enhancement in almost the whole axial skeleton. Complete blood cell count revealed the following: white blood cells, 8.82×109/L; hemoglobin, 132 g/L; and platelets, 159×109/L. Blasts comprised 9% of white blood cells in peripheral blood smear. Bone marrow study confirmed relapse with blasts occupying 84.6% of total nucleated cells and with an immunophenotype similar to that from the initial diagnosis (Fig. 1B, Tables 1 and 2). Genetic analysis with bone marrow aspirate at relapse showed some relevant mutations. G-banding karyotyping revealed 47,XX,+del(1)(p13),t(9;22)(q34;q11.2)[17]/46,XX[8]. RT-PCR and targeted RNA fusion panel confirmed the presence of e1a2-type BCR::ABL1 rearrangement. NGS panel targeting 531 genes related to hematologic malignancy revealed multiple exon deletions in Ikaros family zinc finger 1 (IKZF1; exons 4–7) and retinoblastoma 1 (RB1; exons 18–27). NGS also showed the duplication of chromosome 1q, which is consistent with the karyotyping results (Fig. 3). IGH/IGK gene clonality assay showed clones with the same gene combinations as those at diagnosis (Fig. 2).
None of the genetic alterations at relapse had been captured at diagnosis. The test data at diagnosis were retrospectively reviewed. Karyotyping could not be further verified because the remnant data were only for the initially reviewed 20 cells. However, NGS data showed that some of the mutations at relapse were present even at the initial diagnosis. The deletion of IKZF1 and the duplication of chromosome 1q were too subtle to be reliably captured with routine NGS pipelines but could be observed by the manual inspection of normalized depth plots (Fig. 3). The subtleness could be explained by the low proportion of blasts at the diagnosis.
After reporting the mutations, including BCR::ABL1 rearrangement, imatinib was added to the patient’s chemotherapy regimen. The patient achieved morphologic remission in December 2022. Quantitative polymerase chain reaction for BCR::ABL1 rearrangement converted to negative in February 2023, and the patient received allogeneic hematopoietic stem cell transplantation in May 2023. Although some MRD is suspected from IGH/IGK gene clonality assay, with one IGK clone being 0.003% at the latest assessment, the patient has not relapsed as of the latest follow-up in August 24, 2023 (Fig. 2).
DISCUSSION
The reliable detection of genetic aberrations even in cases of low tumor fraction is an important issue in acute leukemia. Although a diagnosis of ALL is generally not recommended if the bone marrow blast percentage is less than 20%, there is no definite lower limit [1]. A diagnosis of ALL is occasionally given even under a low blast percentage if clinical and laboratory evidence indicates the disease. For AML, there were already some mutations, such as runt-related transcription factor 1 (RUNX1)–RUNX1 partner transcriptional co-repressor 1 (RUNX1T1) rearrangement, that justified the diagnosis of AML even with a blast percentage of less than 20% [2]. The recently revised diagnostic criteria went even further. The fifth edition of the World Health Organization classification removed the 20% blast cutoff for AML with any defining genetic abnormalities, except AML with BCR::ABL1 rearrangement and AML with CCAAT enhancer binding protein alpha (CEBPA) mutation [3]. The International Consensus Classification also lowered the blast percentage cutoff to 10% for many AML-defining genetic aberrations [4]. Under these changes, there will be more cases of acute leukemia with a low fraction of blasts in the bone marrow. These cases will be more prone to false negative results, and the omission of defining genetic aberrations may even lead to diagnoses other than acute leukemia. Therefore, genetic analyses should be more sensitive to catch relevant mutations even in cases of low tumor fraction.
In our case, the proportion of blasts in the bone marrow at initial diagnosis was below 10%. A retrospective review revealed a previously hidden deletion of IKZF1 and a duplication of chromosome 1q, which are consistent with those detected at relapse. RB1 deletion was not observed from the manual inspection of normalized depth plots (Fig. 3). It could have been hidden in even more minor subclones at diagnosis or acquired at relapse; this situation is very rare but has been previously reported [5]. There was no definite evidence of BCR::ABL1 rearrangement at diagnosis. However, from karyotyping at relapse, the Philadelphia chromosome was present in all cells with a partial duplication of chromosome 1, thus lowering the possibility of the late acquisition of the rearrangement. If the rearrangement had been present from diagnosis, a more sensitive genetic analysis might have led to the earlier detection of the rearrangement, thus facilitating administration of a more suitable treatment and the prevention of relapse. This indicates that for acute leukemia with low tumor fraction, efforts are needed to prevent false negative assessments and to ensure that genetic mutations present in tiny proportions are sensitively captured.
Karyotyping is a conventional method for detecting chromosomal duplications, deletions, and rearrangements, which constitute a critical part of relevant mutations in acute leukemia. However, as shown in this case, its sensitivity is low, particularly for low tumor fraction samples. To prevent tumor cells from being overlooked because of the presence of normal cells, reviewing more than 20 cells may be considered. Furthermore, if at least one cell with abnormality is observed, a further confirmation of the mutation will be appropriate rather than simply dismissing it as a false finding.
Other than karyotyping, assays with higher sensitivity need to be more actively applied. Fluorescent in situ hybridization can have a limit of detection of 5% or lower depending on the specific technique, and RT-PCR can detect one aberrant cell among 100,000 normal cells [6]. Concerning NGS technology, more progress is needed to reliably detect low-fraction copy number variants and differentiate them from false positives. However, for single-nucleotide variants and short indels, efforts have been made to lower the limit of detection to less than 1% [7, 8]. NGS can also be applied to RNA-based fusion gene detection with a maximum sensitivity of 1 per 10,000 cells [9]. Even if some genetic tests are inevitably prone to false negative results, the active application of these sensitive assays can lower the combined false negative rate.
The IGH/IGK gene clonality assay is a genetic test that does not target diagnosis-defining mutations but aids in MRD follow-up. An uncommon finding in this case was that even when IGH clonality was converted to negative, IGK clonality still indicated MRD. In a previous study, cases where IGH clonality decreased beyond 10-6 with remaining IGK or immunoglobulin lambda locus (IGL) clonality at the end of consolidation comprised 10.6% of patients with pediatric B-ALL [10]. IGK or IGL MRD without IGH MRD was not correlated with three-year event-free survival in the study. However, the patient in the current case relapsed approximately 3.5 years after initial diagnosis, thus possibly indicating the need for follow-up of not only IGH but also IGK/IGL clonality for long-term prognostic predictions.
This study reported a case of acute leukemia where low tumor fraction at diagnosis led to false negative genetic assessments. More sensitive assays would have prevented false negative results and may have led to more adequate treatment and better outcomes. This case indicates that efforts for higher sensitivity need to be made, particularly for acute leukemia with low tumor fraction.
 XML Download
XML Download