

 PDF
PDF Citation
Citation Print
Print



서 론
검사법 평가(method evaluation)는 임상 검사실(clinical laboratory)의 질 보증(quality assurance)을 위한 기초적이고 필수적인 과정이다[1-5]. 일반적으로 신규 검사나 장비 도입 시에 각 검사실에서는 초기 평가절차를 시행하는데, 많은 검사실에서는 자체 운영지침서를 통해 초기 도입평가 과정과 절차에 대해 명시하고 이에 맞추어 초기 평가를 시행하고 있으나, 아직 평가지침에 대한 일치된 지침(guideline)이 존재하지 않고 각 기관마다 상이한 것이 현실이다[1, 4-6]. 또한 이는 검사실자체개발검사(laboratory developed test, LDT) 평가에서도 마찬가지라서 현재로서는 LDT 평가절차는 각 검사실 자체의 지침서에 의존하고 있는 실정이다. 현재 진단검사의학재단 우수검사실 신임인증 심사점검표에서는 “신규 검사나 장비 도입 시 또는 검사 방법이나 시약의 큰 변화가 있는 경우 검사의 성능을 검증하는가?”라는 문항을 통해 신규 검사법 평가 여부를 평가하고는 있으나, 이에 대한 세부 지침 및 하위항목이 없어서 해당 문항에 대한 평가가 평가자의 주관에 따라 달라질 수도 있고, 수검 기관에서도 이에 대한 대비가 어려운 점이 있다는 한계가 있다[7].
이에 본 연구에서는 신규 검사나 장비 도입 시 또는 검사 방법이나 시약의 큰 변화가 있는 경우에서의 신규 검사법 평가절차 각 과정 및 절차가 검사실마다 실제 어떠한 방식으로 이루어지고 있는지 설문조사를 통해 현황을 확인하였다. 이를 통해 각 검사실 또는 기관에서의 검사법 평가절차에서의 실태 및 문제점을 파악하고 분석하여, 추후 검사법 평가절차에 대한 표준화된(standardized) 국내 지침 마련을 위한 기초 자료를 제시하고자 하였다.
재료 및 방법
국내 의료기관 및 전문수탁의료기관(referral medical laboratories)에 근무하는 진단검사의학과 전문의들을 대상으로 설문조사를 시행하였다. 2023년 6월 27일부터 8월 9일까지 총 243명에게 이메일을 발송하여 온라인 설문조사를 시행하였다. 특정 온라인 플랫폼을 사용하지는 않고 설문조사 문항 및 답가지를 엑셀 파일에 수록하고 이를 이메일에 첨부하였다. 이메일을 수신한 전문의는 자발적으로 연구 참여를 희망할 경우 엑셀 파일에서 답변을 콤보 박스에 체크(한 문항에 복수응답 가능)하여 이메일로 회신하도록 하였다. 한 기관에 전문의가 여러 명인 경우 이 중에서 임상화학 분과를 담당하는 전문의에게 보냈으며, 해당 기간 동안 총 243명의 전문의에게 이메일을 발송하여 이 중 60명(24.7%)에게서 회신을 받았다.
설문 참여 기관의 기본 정보로 기관의 소재 지역, 종별 형태, 병상 수, 연간 검사 건수 등을 조사하였고, 각 기관에서 검사법 평가를 위해 사용하는 방법 및 통계 프로그램 역시 조사하였다. 검사법 평가에 대한 세부 설문은 총 32문항으로 4개의 부분으로 나누어 이루어졌다. 1부는 정밀도(precision) 분야로서 정밀도 8문항, 2부는 직선성(linearity) 분야로서 직선성 6문항 및 측정가능구간(analytical measuring interval) 2문항이었고, 3부는 비교 평가 분야로서 방법 간 비교(method comparison) 4문항, 시약 lot 간 평행 검사(parallel test) 3문항, 동등성 검사(comparability test) 1문항으로 구성하였고, 4부는 기타 분야로서 측정능(detection capability) 3문항, 잔효(carryover) 1문항, 참고구간(reference interval) 1문항, 정성검사(qualitative test) 성능평가 3문항으로 구성하였다.
결 과
1. 설문 참여기관 현황
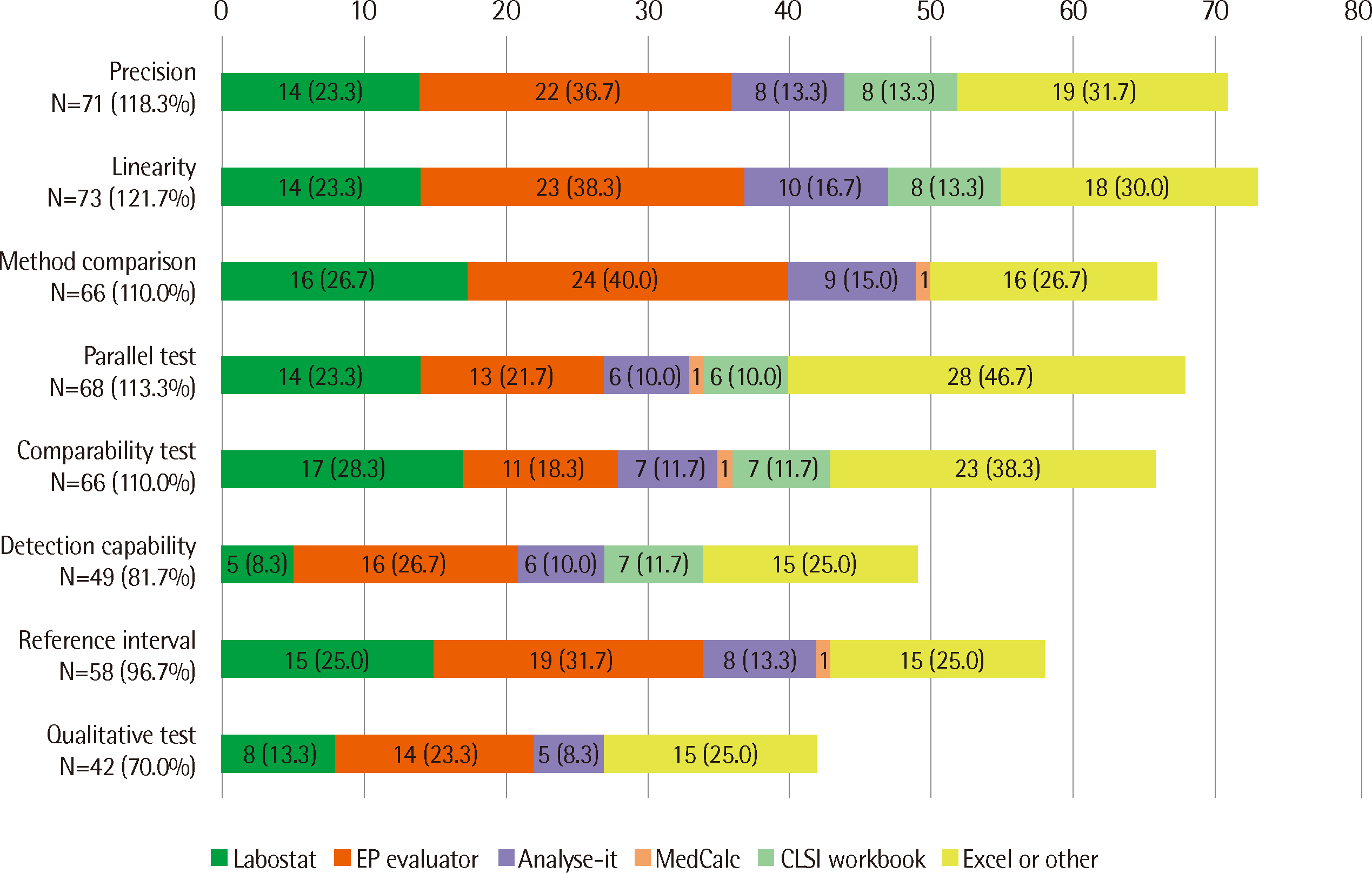
본 연구에 참여한 각 기관의 소재지는 수도권 42기관(70.0%), 영남권과 충청권 각 5기관(8.3%), 호남권과 강원권 각 4기관(6.7%)의 순이었고 제주권에서는 참여 기관이 없었다. 의료기관 종별로는 상급종합병원 21기관(35.0%), 종합병원 및 병원급 의료기관 34기관(56.7%), 의원급 의료기관 1기관(1.7%), 전문수탁의료기관 4기관(6.7%) 등이었으며, 의료기관의 병상 수는 500병상 미만 14기관(23.3%), 500–1,000병상 32기관(53.3%), 1,000–2,000병상 7기관(11.7%), 2,000병상 이상 2기관(3.3%)이었다. 전체 응답기관 중 31기관(51.7%)에서는 외부에서 의뢰되는 검체에 대한 수탁검사를 시행하고 있다고 응답하였고, 57기관(95.0%)은 진단검사의학재단 우수검사실 신임인증을 받는 기관이었다. 신규 검사법 도입 평가 결과에 대해서는 기기 혹은 시약회사 학술부에서 통계처리한 후에 각 기관의 전문의가 검토하는 경우가 48기관(80.0%), 검사실 실무자가 통계처리한 후에 전문의가 검토하는 경우가 20기관(33.3%), 전문의가 직접 통계처리 및 검토하는 경우가 10기관(16.7%)이었다. 검사법 평가에 사용하는 통계 방법 및 프로그램은 Fig. 1과 같다(복수응답 있음).
2. 정밀도
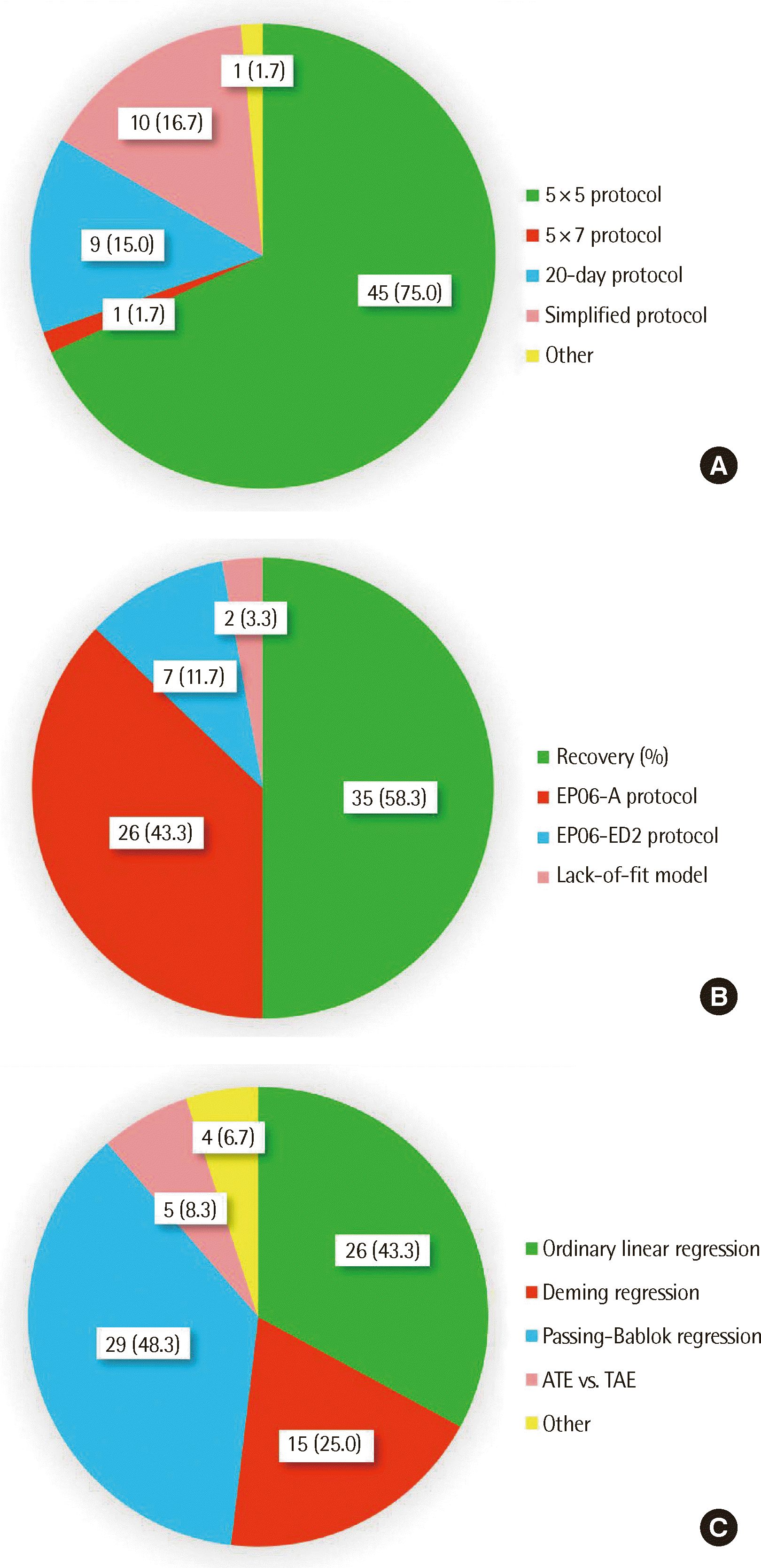
정밀도 평가 설문에 대한 답변을 Table 1에 정리하였다. 설문조사 참여 기관 중 57기관(95.0%)에서는 정밀도 평가를 위한 최신 지침인 CLSI (Clinical and Laboratory Standards Institute) EP15-A3 [8]에 대해 안다고 응답하였으나 실제로 5×5 프로토콜(protocol) (하루에 1번 시행, 시행당 5회 반복측정을 5일간 시행하여 각 정량 값(level)당 총 25회 측정)을 사용하는 기관은 45기관(75.0%)이었다(Fig. 2A). EP05-A3 [9]에 제시된 20일 프로토콜(하루에 2번 시행, 시행당 2회 반복측정을 20일간 시행하여 각 레벨당 총 80회 측정)을 사용하는 기관은 10기관(16.7%)이었다. 반복측정재현성(repeatability)에 대해서는 21기관(35.0%)에서 별도로 평가한다고 응답하였으나, 기시행한 정밀도 평가 데이터를 기반으로 계산하고 추가적인 반복측정재현성 평가를 시행하지 않는다는 경우가 34기관(56.7%)으로 더 많았다. 시행 간 비정밀도(between-run imprecision), 총검사실 비정밀도(total-laboratory imprecision)를 따로 계산하지 않고 전체 정밀도 측정 데이터의 %변이계수(coefficient of variation)만으로 평가한다는 기관은 31기관(51.7%)이었다. 이상치(outlier)에 대해서는 따로 평가하지 않고 모든 정밀도 평가 데이터를 활용한다는 기관이 33기관(55.0%)이었으며, 전체 정량 값에서 최대 1개까지 제거하는 기관이 5기관(8.3%), 2개까지 제거한다는 기관이 18기관(30.0%)이었다. 정밀도 평가를 적합(acceptable)으로 평가하는 기준은 제조사 제시 %CV 41기관(68.3%), 문헌에 나오는 %CV 16기관(26.7%), 검사실 자체 설정 %CV 19기관(31.7%)이었다. 정밀도 평가 결과가 부적합(unacceptable)으로 판정되는 경우 EP15-A3 [8]에 의거하여 검정상한(upper verification limit, UVL)을 확장시켜 평가하는 기관은 21기관(35.0%)이었으나, 24기관(40.0%)에서는 추가 검정 과정 없이 곧바로 부적합 판정을 내린다고 응답하였다.
3. 직선성
직선성 평가 설문에 대한 답변을 Table 2에 정리하였다. 52기관(86.7%)에서 직선성 평가를 위한 최신 지침인 EP06-ED2 [10]에 대해 안다고 응답하였으나 실제로 개정 프로토콜을 사용하는 기관은 7기관(11.7%)이었고, EP06-A [11]에 따른 이전 프로토콜을 사용하는 기관이 26기관(43.3%), 기대치(expected value)와 실측치(measured value) 간의 %회수율(recovery) 사용이 35기관(58.3%)이었다(Fig. 2B). 직선성 평가의 기준은 생물학적 변이(biological variation)를 고려하지 않고 모든 항목에 대해 ±7%, ±10% 등 일괄적으로 설정한 경우가 31기관(51.7%)으로 가장 많았다(Fig. 3). 최저 정량 값에 대한 판정 기준으로는 최저 정량 값에서만 기준을 벗어나고 다른 모든 정량 값에서 모두 기준을 만족하는 경우 전체 구간에서 직선성을 만족한 것으로 간주하는 경우가 21기관(35.0%), 최저 정량 값에 대해서만 %기준을 넓혀서 별도로 적용하는 경우가 15기관(25.0%), 최저 정량 값에 대해서는 %기준이 아닌 절대값 기준을 적용하는 경우가 21기관(35.0%)이었다.
희석배수(dilution factor)에 대한 최신 지침인 EP34-ED1 [12]를 안다고 응답한 기관은 46기관(76.7%)이었으나 실제로 해당 프로토콜을 적용하는 기관은 13기관(21.7%)이었고, 제조사에서 제시하는 희석배수를 별도의 검정 없이 그대로 사용한다고 응답한 경우가 32기관(53.3%)으로 가장 많았다.
4. 방법 간 비교
방법 간 비교 평가 설문에 대한 답변을 Table 3에 정리하였다. 방법 간 비교 프로토콜로는 최소제곱 선형회귀(least square linear regression)분석(26기관, 43.3%)과 Passing-Bablok 회귀분석(29기관, 48.3%)을 가장 많이 사용하고 있었으며(Fig. 2C), 적합 판정에 대한 기준은 기관마다 다양하였다. 방법 간 비교 평가의 기준 역시 모든 항목에 대해 일괄적으로 설정한 경우가 29기관(48.3%)으로 가장 많았고(Fig. 3), 사용되는 환자검체 개수는 40개인 경우가 39기관(65.0%)으로 가장 많았다. 시약 lot 번호 변경 시 시행하는 평행검사에 대해서는 기존 lot과 새로운 lot으로 측정하는 검체 수가 2–5개인 경우가 41기관(68.3%)으로 가장 많았고, 평가 기준 역시 모든 항목에 대해 일괄적으로 설정한 경우가 31기관(51.7%)으로 가장 많았다(Fig. 3). 평행검사 결과 평가에 있어서 EP26-A [13]에서 제시된 임계차이(critical difference)를 사용하지 않고 %차이(difference)를 사용한다고 응답한 경우가 51기관(85.0%)으로 가장 많았다. 동일 검사실 내 같은 종목을 2가지 이상의 기기로 검사하는 경우의 정기적인 기기 평가는 이전의 C54-A [14] 혹은 최신 EP31-A-IR [15] 프로토콜이 아닌, 매일 1개 혹은 약간의 검체를 측정하고 이를 누적하여 평가하는 경우가 35기관(58.3%), 방법 간 비교평가와 동일한 프로토콜을 사용하는 경우가 24기관(40.0%)이었다.
5. 기타 항목
기타 항목 설문에 대한 답변을 Table 4에 정리하였다. 측정능에 있어서 공시료 측정한계(limit of blank), 검출한계(limit of detection), 정량한계(limit of quantitation)를 검증/검정하는 기관이 25기관(41.7%)이었고, 이 중에서 정량한계 검증/검정 기준은 총오차(total error) 9기관(15.0%), %변이계수 16기관(58.3%)이었다. 잔효 평가의 기준은 고농도 검체 4회 연속 측정 후 곧이어 저농도 검체 4회 연속 측정하는 기관이 49기관(81.7%)이었다. 참고구간에 있어서는 모든 기관에서 제조사 제시 참고구간을 20개의 건강인 검체를 이용하여 평가한 후 전이(transference)하여 사용하였는데, 추가적으로 참고구간을 설정(establish)한다고 응답한 18기관(30.0%)이었다. 정성검사 성능평가에서는 각 정량 값당 2–3개 검체를 사용하는 경우가 22기관(36.7%), 5개 검체를 사용하는 기관이 16기관(26.7%)으로 가장 많았고, 부적합 판정이 나올 경우 처음 평가한 검체 수만큼 추가로 검사하여 재평가한다고 응답한 기관이 24기관(40.0%)으로 가장 많았다.
고 찰
진단검사의학(laboratory medicine)은 임상적 진단은 물론이고 질병의 예방과 감시(monitoring), 치료에 대한 반응 및 예후 평가, 환자 맞춤형 치료 모델 제공 등에 있어서 중요성이 큰 의학 분야이며[2, 16], 진단검사의학의 지속적인 발전과 함께 미래의 보건의료에서 진단검사의학의 역할은 더욱 증대될 것이다[17]. 이러한 관점에서 임상검사실에서의 검사법 평가[1-5] 및 체외진단기기(in vitro diagnostic device)의 도입 시 평가절차는 그 중요성이 크다[4, 5, 18, 19]. 검사법 평가는 검증(validation)과 검정(verification)으로 나누어 볼 수 있다. 측정학지침합동위원회(Joint Committee for Guides in Metrology) [20]에 따르면 검증은 측정시스템의 명시된 요구 사항이 사용 목적에 적합한지를 평가하는 것으로, 검정은 측정시스템의 목표 성능 특성이나 법적 요구사항이 달성되었는지를 평가하는 것으로 정의하고 있다. CLSI EP19-ED2 [19]에서는 검증은 제조사가 측정시스템의 분석적, 임상적 성능 특성을 설정(establishment) 및 확정(confirmation)하는 과정으로, 검정은 최종 사용자(end-user)인 검사실에서 측정시스템의 수행능 특성이 제조사 설정 목표(claim)를 충족하는지를 확인하는 과정으로 정의하고 있다. 국제표준기구(International Organization of Standardization, ISO)의 임상검사실의 질 관리 지침인 ISO 15189 [21]에서는 비표준 검사법, LDT, 특정 측정시스템을 명시된 사용 목적 이외에 사용할 경우, 검증이 이미 된 측정시스템에 변경사항이 생기는 경우 등에 있어서 검증이 필요하다고 밝히고 있고, 검정에 대해서는 반드시 검증이 필요한 경우가 아닌 경우 혹은 이전에 수행한 평가가 불충분한 경우 최소한의 검정은 필요하고, 동일 기관에서 동일 종목에 대해 두 개 이상의 측정시스템을 사용할 경우 각 개별 측정시스템에 대한 검정이 각각 필요하며, 검증 및 검정의 전 과정, 지침 및 결과는 반드시 문서화해야 한다고 밝히고 있다. 또한 검증 및 검정에 필수적인 요소를 정밀도, 직선성, 정확도(accuracy), 측정한계, 측정범위, 잔효, 간섭(interference), 분석적 민감도와 특이도 등으로 정의하고 있다[1, 4, 18, 21, 22].
검사법 평가 절차에 대한 국내 현황에 대해서는 이전 문헌이 존재하지 않아 정확한 파악은 힘들지만 국외의 경우와 마찬가지로 각 검사실 간의 차이가 클 것으로 추정된다. 그동안 진단검사의학재단은 물론 대한진단검사의학회 및 대한임상화학회에서도 관련 내용을 교육한 바 있으나 이에 대한 표준화된 지침은 아직까지 제공된 바 없다. 또한 이전의 국내 교과서에서는 관련 내용을 다루지 않았고, 2021년에 발간된 진단검사의학 제6판[23] 및 진단검사의학 실무핸드북 제1판[24]에 와서야 처음 검사법 평가에 대한 내용이 수록되었다. 이에 본 연구에서는 국내에서의 검사법 평가 절차 및 방법의 일치화 및 표준화를 위한 기초 자료를 마련하기 위하여 국내 임상 검사실을 대상으로 설문조사를 시행하였다.
앞서 밝혔듯이 설문조사 참여기관의 분포는 수도권이 70.0%를 차지하였는데, 처음에 설문조사 이메일을 보낸 243기관 중에는 수도권이 130기관(53.5%)이었으나, 지방 소재 기관의 참여율이 수도권 소재 기관보다 상대적으로 낮았다. 의료기관 종별로 살펴보았을 때에도 상급종합병원이 35.0%를 차지하였는데, 이는 상급종합병원의 경우 검사법 평가를 비롯한 검사실의 질 관리 전반에 관심과 노력을 더욱 많이 기울이는 것은 물론 임상화학 분야에 대한 관심도가 높고 학회 참석을 통해 검사법 평가에 대한 사전 인지도가 높았던 것이 그 이유로 생각되며, 이로 인해 본 설문조사의 결과에서 나타난 응답 수와 비율은 실제 국내 임상 검사실의 평균보다는 다소 높을 것으로 생각된다. 신규 검사법 도입 평가 결과에 대해서는 기기 혹은 시약회사 학술부에서 통계처리한 후에 각 기관의 전문의가 검토하는 경우가 80.0%를 차지하였는데, 이는 전문의와 임상병리사 이외에도 기기 혹은 시약회사 직원들을 대상으로 하는 검사법 평가 교육의 필요성을 역설한다. 검사법 평가를 위한 통계 프로그램의 경우 진단검사의학재단에서 개발한 표준통계프로그램인 Labostat (Laboratory Medicine Foundation, Seoul, Korea)은 검출능과 정성검사를 제외한 종목에서 EP Evaluator (Data Innovations, Colchester, VT, USA) 바로 다음인 20%대의 이용률을 보였고, 한편으로 Analyse-it (Analyse-it Software, Ltd., Leeds, UK)의 이용률이 낮은 것은 고가의 구매 비용으로 생각되며 EP Evaluator 의 이용률이 예상보다 높은 것은 앞선 문항에서 밝힌 바와 같이 기기 혹은 시약회사 학술부에서의 EP evaluator 사용률이 상대적으로 높기 때문으로 생각된다. 한편으로는 통계 프로그램 이외에도 자체적으로 Microsoft Excel (Microsoft Corporation, Redmond, WA, USA)에 수식을 입력하여 통계 처리하는 경우도 많았다. CLSI에서는 2020년대부터 기존 CLSI 지침에 대한 IG (implementation guideline)와 WB (workbook)를 회원들을 대상으로 제공하고 있는데, 현재 정밀도, 직선성, 희석배수, 검출능, 총분석오차, 평행검사, 동등성 평가 등에 대해 워크북(Microsoft Excel 파일 형태)을 제공하고 있고 2023년 8월 현재 방법 간 비교, 참고구간, 정성검사 평가 등에 대한 워크북은 제공하고 있지 않다. 아직 CLSI 워크북이 출시된 지 많은 시간이 지나지 않은 데다가 CLSI eCLIPSE 홈페이지에 가입한 회원들만 다운로드가 가능하기에 CLSI 워크북의 이용률이 아직은 저조한 것으로 보인다.
정밀도 평가에서는 응답기관의 75.0%에서 EP15-A3 [8]에 의거한 5×5 프로토콜을 사용하여 국외 설문조사 결과[5, 6]에 비해 CLSI 지침을 적용하는 비율이 높은 것으로 나타났다. 반복측정재현성의 경우 별도로 측정하지 않는 경우가 많은 것으로 나타났는데, 이는 통계 프로그램을 통해 기존 프로토콜 데이터를 가지고 반복측정 비정밀도를 산출할 수 있기 때문으로 보인다. 이상치에 있어서는 EP15-A3에서는 한 정량 값에서 최대 1개, 전체 정량 값에서 최대 2개까지 제거가 가능하다고 밝히고 있는데[8], 전체 응답기관의 95.0%에서 EP15-A3에 대해 안다고 답변한 것을 고려해 보았을 때에, 정도관리물질을 이용한 정밀도 평가에서 이상치가 잘 발견되지 않아서 제거하지 않았거나 혹은 기관의 전문의가 이상치를 제거하지 않고 모든 데이터를 바탕으로 정밀도를 평가해야 한다고 내부 지침을 세웠을 가능성이 있다. 정밀도 평가결과의 판정 기준은 제조사 제시 %CV가 68.3%이었는데, 이는 국외 설문조사 결과에서 나타난 71% [5]와 60% [6]와 비슷하였다. 한편으로 5×5 프로토콜을 통해 시행 간 비정밀도, 총검사실 비정밀도를 구할 수 있는데 절반이 넘는(51.7%) 검사실에서는 시행에 따른 요소를 고려하지 않고 25회 전체 측정치 분포에 대한 %CV로만 판정하고 있어, 이에 대한 교육이 필요할 것으로 생각된다. 판정 기준을 만족하지 못할 경우의 조치에서는 무려 40.0%의 기관에서 추가적인 조치 없이 부적합 처리를 한다고 하였는데, 국외 사례에서는 28%의 경우 허용 가능한 편차인지를 살펴보고 36%에서는 추가 검사를 통해 재평가한다고 응답한 만큼[6] 국내에서도 EP15-A3 [8]에 제시된 검정상한을 이용하는 방법 및 기타 조치에 대한 교육 또한 필요할 것으로 보인다.
직선성 평가에서는 응답기관의 86.7%에서 EP06-ED2 [10]에 대해 안다고 답변하였으나 실제로는 기대치와 실측치 간의 회수율을 토대로 평가하는 경우가 많았다. 이는 EP06-A [11]나 EP06-ED2 [10] 프로토콜을 적용하기 위해서는 별도의 통계 프로그램이 필요하지만 회수율의 경우에는 Excel 프로그램에 수식을 대입하기만 하면 되기 때문인 것으로 보인다. 직선성 평가결과의 판정 기준은 모든 항목에 대해 ±7%, ±10% 등 일괄적으로 설정한 경우가 51.7%으로 가장 많았지만, 생물학적 변이에 대한 학회 차원의 주기적인 교육 덕분에 생물학적 변이를 참고하는 경우도 38.3%로 비교적 높게 나타났다. 이상치가 발견되었을 때의 조치와 최저 정량 값에서의 판정 기준을 별도로 적용하는지의 여부는 기관별로 다양하였다. 희석배수에 있어서는 절반이 넘는(53.3%) 기관에서 제조사가 제시하는 희석배수를 별도의 검정 과정 없이 그대로 사용한다고 하는데, 이는 확장측정구간(extended measuring interval) 역시 제조사 제시 구간을 그대로 사용함을 의미한다. 이에 희석배수 검정에 대한 각 임상 검사실, 기기 및 시약회사의 관심 증대가 요구된다.
방법 간 비교의 프로토콜과 판정 기준은 기관별로 다양하였다. 방법 간 비교에 사용하는 환자 검체 개수는 40개라고 답변한 기관이 65.0%로 가장 많았다. 국외 설문조사 연구에서는 20, 40, 50, 100개 등으로 다양한 분포를 보이는 점[6]과 비교해 보았을 때, 국내의 경우에는 CLSI 지침을 참고하는 경우가 많다 보니 EP09-ED3 [25]에서 언급한 검정을 위한 최소 권장 개수인 40개를 지키는 경우가 많은 것으로 보인다. 시약의 lot이 변경되었을 때의 평행 검사와 단일 기관에서 같은 종목을 2가지 이상의 기기로 평가하는 경우의 동등성 비교에 대해서는 각각 해당 지침인 EP26-A [13]와 EP31-A-IR [15]에 의거하여 평가하는 경우가 많지 않고, 약간의 환자 검체를 이용하여 측정한 후 %차이로만 계산하는 경향이 컸다. Labostat을 비롯하여 많은 검사법 관련 통계 프로그램에서 이들에 대해 다루고 있으나, 많은 검사실에서는 편의성 때문에 Excel 프로그램을 이용하여 %차이만 계산하는 경우가 많은 것으로 보인다. 해당 파트에서 EP26-A [13]와 EP31-A-IR [15] 지침을 알고 있는지에 대한 문항을 수록하지 못하여 실제 임상 검사실에서 이들 지침에 대해 어느정도 알고 있는지에 대한 파악은 하지 못하였다.
LoB, LoD, LoQ 등의 측정능에 대해서는 많은 임상 검사실에서 별도의 검증이나 검정을 하지 않는 것으로 나타났고, 잔효 평가의 기준은 고농도 검체 4회 연속 측정 후 곧이어 저농도 검체 4회 연속 측정하는 경우가 가장 많았다. 참고구간에 있어서는 전반적으로 EP28-A3c [26]에 의거하여 제조사에서 제시하는 참고치를 잘 전이해서 사용하는 것으로 나타났다. 정성검사 성능평가에 있어서는 EP12-A2 [27]에서는 C50-20%와 C50+20% 물질을 각각 20개 사용할 경우 신뢰도가 71.8%, 각각 40개 사용할 경우 신뢰도가 85.8%를 보인다고 밝히고 있는데, 실제 임상 검사실에서는 연 2회 시행하는 정성검사 성능평가에 있어서 평가 물질 확보가 쉽지 않다보니 각 정량 값당 5개 이하의 검체를 사용하는 경우가 많았고, 부적합 판정을 보였을 때의 조치 방법도 기관별로 상이하였다.
정확도에 대해서는 특수한 상황이 아닌 한, 일반적인 임상 검사실에서는 기존 검사법을 참조 방법으로 간주하고 신규 도입하는 검사법을 평가 검사법으로 하여 방법 간 비교로 대체하는 경우가 많으며, 참조표준물질(reference standard material)을 이용한 정확도 평가 및 불확도(uncertainty) 평가는 잘 시행하지 않아 본 설문의 문항에는 포함시키지 않았다. 추가적으로 여러 지침들에서 간섭현상에 대한 평가를 포함할 것을 명시하고 있는데[1, 4, 18, 21, 22], 많은 기관에서 추가적인 평가 없이 제조사에서 제시하는 간섭물질 기준을 그대로 적용할 것으로 판단되어서 이 역시 본 설문에는 포함하지 않았다.
본 연구는 검사법 평가에 대한 국내 임상 검사실의 지침 및 현황에 대해 조사한 최초의 연구로서, 국내 검사실 실정에 맞는 검사법 평가절차 항목 및 문항에 대한 일치화의 필요성을 환기하는 것을 1차적 목적으로 하고 있으며, 더 나아가서는 검사법 평가절차에 대한 각 검사실의 평가절차서를 마련하는 데에 아웃라인(outline)을 제공하고 참고자료로 활용함에도 그 목적이 있다. 본 연구를 통해 나타난 국내 임상 검사실의 검사법 평가 현황을 바탕으로 향후 더 많은 후속 연구가 진행되어 국내 검사실의 검사법 평가 지침 및 절차에 대해 참고할 만한 표준화 혹은 일치화된 지침이 마련되어야 할 것이다. 더 나아가서는 본 연구를 비롯한 연구결과들이 검사법 평가절차에 대한 진단검사의학재단 우수검사실 신임인증 심사점검표에 반영되어 각 기관에서 우수검사실 신임인증을 정확하고 수월하게 준비할 수 있도록 도움을 주며, 각 기관에서 통일되고 CLSI 지침에 부합한 방식으로 신규 검사방법을 평가할 수 있도록 하여 궁극적으로는 각 기관의 검사실의 질 관리 수준을 향상하여야 할 것이다.
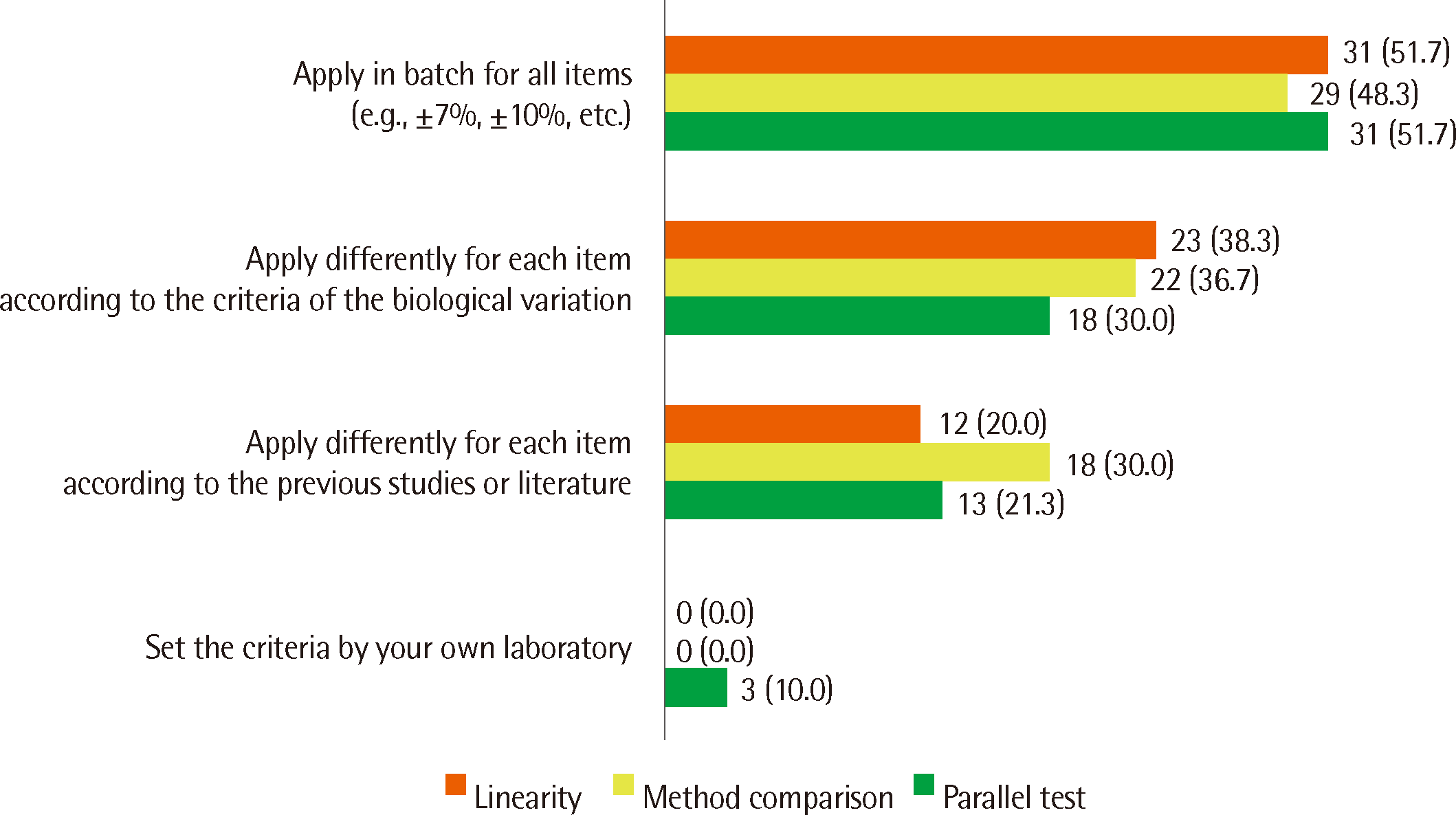
 XML Download
XML Download