

 PDF
PDF Citation
Citation Print
Print



서 론
최근, 순환 종양 DNA (circulating tumor DNA, ctDNA)는 조기 진단, 표적 치료 마커 탐색, 치료 반응 모니터링, 예후 평가 및 미세잔존질환(minimal residual disease) 탐지를 위한 최소 침습 도구로 관심을 받고 있다[1-3]. ctDNA는 세포자멸사(apoptosis) 및 괴사(necrosis) 과정에 있는 종양 세포 또는 살아있는 종양 세포로부터 유래하는 세포유리 DNA (cell-free DNA, cfDNA)의 일종이다[4]. 일반적인 cfDNA는 크기가 약 167 base pairs인 반면, ctDNA는 더 많이 분절화되어 그 크기가 더 작은 것으로 보고되었다[5]. ctDNA의 검출은 주로 cfDNA에서 체세포 변이(somatic variant)를 확인함으로써 검출한다. 본 지침의 목적은 현재 최신 근거를 기반으로 하여 ctDNA의 검사 전단계, 검사 단계, 해석 및 결과보고에 관하여 권고하고자 함에 있다.
가이드라인 개발 과정
권고 개발의 원칙은 임상 검사실에서의 ctDNA검사 수행에 대해 근거에 기반한 합의를 도출하는 것이다. 권고 개발은 기본적으로 한국임상진료지침 개발을 위한 수용개작 과정 버전 2.0 (Adaptation Process for Developing Korean Clinical Practice Guidelines Ver. 2.0)의 방법을 따랐으며[6], 이 작업에는 1) 대한진단검사의학회(Korean Society for Laboratory Medicine)의 진료지침위원회; 2) 근거에 기반한 권고를 검토하고 의견를 제공한 6명의 진단검사의학과 전문의로 구성된 전문가 패널 두 그룹이 참여했다. 진료지침위원회는 먼저 본 지침의 범위를 정하고, 그 후 PubMed, KoreaMed, 그리고 Google Scholar에서 영어 그리고 한국어로 논문을 각각 검색하였다.
검사전 단계의 경우, 2000년 1월부터 2022년 7월까지 발표된 논문 중 특정 주제어를 바탕으로 검색을 하였고, “cell-free DNA” AND “pre-analytical”, “ctDNA” AND “pre-analytical”, and “circulating DNA” AND “pre-analytical”의 주제어 조합이 사용되었다. 검색된 268편의 논문 중, 진료지침위원회는 그 논문들의 제목과 초록을 검토하여, 중복 기록을 제외하고 인간을 대상으로 시행한ctDNA 검사의 검사전단계에 관련된 77개의 논문을 남겨두었다. 해당 논문들의 주요 내용은 그 후 SIGN과 K-AGREE-II 평가도구에 준하여 검토되었는데[6], 적절하게 설계되고 편향의 위험이 낮은 연구가 본 지침에 포함되었다. 최종적으로, 검사전 단계에 대한 논문 중 본 지침에 포함된 논문은 27편이다(Table 1).
검사 기술을 포함한 검사 단계 측면은 PubMed에서 “analytical” AND “ctDNA”, 그리고 “analytical” AND “cell-free DNA”의 주제어 조합을 사용하여 검색되었다. 검색된 527 편의 논문 중, 그 논문들의 제목과 초록을 검토하여, 중복 기록을 제외하고 인간을 대상으로 시행한 ctDNA 검사의 검사 단계에 관련된 23개의 논문을 남겨두었다.
해석 및 결과보고의 경우, PubMed에서(“ctDNA” OR “tumor NGS”) AND (“reporting” OR “interpretation”) AND (“guideline” OR “consensus” OR “recommendation”)의 조합을 사용하여 검색하였다. 검색된 273편의 논문 중, 그 논문들의 제목과 초록을 검토하여, 중복 기록을 제외하고 인간을 대상으로 시행한 ctDNA 검사의 검사 단계에 관련된 13개의 논문을 남겨두었다.
초안 지침서의 개발 후, 분자진단 분야 전문가 6명으로 이루어진 자문위원회로부터 초안 지침서에 대한 의견을 얻기 위하여, Delphi 방법을 사용하여 질문지가 2회에 걸쳐 발송되었다[7]. 총 17개의 권고안 중, 등급 시스템을 이용하여 첫 번째 조사 내 CV >15%인 6개의 권고안(1 강하게 반대-9 강하게 찬성)이 다시 2차 조사 의견을 위해 발송되었다. 자문위원회의 의견에 따라 전문가 다학제 팀과의 치료약제 선택에 대한 토의에 대한 1개의 권고안이 제거되었고 최종적으로 16개의 권고안이 개발되었다. 근거 수준(Table 1)과 권고의 점수(Table 2)는 과거 임상 적용 지침서와 점수 시스템을 고려하여 개발되었다[8, 9].
검사전 단계
1. 채혈 시점
암환자에서 ctDNA의 농도는 치료에 대한 반응에 따라 달라지기 때문에, ctDNA 분석을 위한 채혈 시점은 검사 목적을 고려하여 신중하게 계획해야 한다[10]. 종양의 진단이나 재발 시에 치료제 선택을 위한 목적일 경우, 수술 및 방사선 치료, 항암화학 치료를 시행하기 전에 채혈을 하는 것이 권장된다. 종양이 치료에 반응하는 시기나 진행하지 않는 시기에 채혈할 경우, ctDNA의 농도가 낮아져서 위음성 결과가 나올 수 있다[10]. 또한, 수술 및 항암화학 요법으로 인한 조직 손상은 정상 cfDNA를 증가시킬 수 있어서, 그 결과 ctDNA의 분율(fraction)이 검사의 검출 한계 미만으로 낮아질 수 있다[10-13]. 따라서, 치료 후 잔존 질환을 확인하고 재발을 예측하기 위한 목적이라면 치료 직후 채혈하는 것은 피해야 한다[10, 11]. 조직 손상의 정도와 회복 시간에 따라, 수술 후 최소 1주에서 2주 후에 채혈을 시행하는 것이 권장된다[10].
2. 채혈 및 검체 보관, 운송
2) 채혈관 및 검체 보관
채혈관에 있는 항응고제는 혈장 분리 전 혈액 응고를 방지한다. 항응고제 중 K2- 혹은 K3-EDTA는 DNA분해효소의 활성을 억제하며, 세포를 용해로부터 안정화 시키고, PCR 반응을 저해하지 않아서 cfDNA 분석에 적합하다[17, 20, 21]. EDTA-함유 채혈관으로 채혈한 후 4–6시간이 지나면 백혈구 용해로 인하여 총 DNA 농도가 증가한다[20-22]. 따라서, EDTA 채혈관을 사용할 경우 정상 DNA로 인한 오염을 막기 위하여 혈장 분리를 가능한 빠르게 시행해야 하며 4–6시간을 넘기지 않도록 해야한다[20-22]. EDTA 채혈관에 혈액을 4–6시간 보관할 경우 상온(18–25°C)이나 4°C 사이에 검체 안정성의 큰 차이가 없는 것으로 보고되어 있다[14, 22]. 하지만 불가피하게 혈장 분리가 6시간 이상 지연될 경우, 하루 정도까지 4°C에서 보관이 가능하다[20-22]. 세포보존 채혈관(cell preservation tube)을 사용할 경우 채혈 후 혈장 분리까지의 시간 간격을 연장할 수 있다[16, 20, 22]. 세포보존 채혈관을 사용할 경우, 제조사의 권고사항에 따라 검체를 처리하고 보관해야한다[22]. 일반적으로, 세포보존 채혈관에 채혈한 혈액은 상온에서 5–7일간 보관할 수 있다[20, 22]. 채혈관에 채혈 후에는 채혈관을 8–10회 정도 위아래로 부드럽게 혼합하여 첨가제와 혈액이 충분히 섞이도록 한다[20, 22].
3. 혈장 분리 및 정도관리, 보관
3) 혈장의 보관
분리한 혈장은 즉시 4°C로 냉각한 후 DNA 추출 시까지 동결 보관하여 핵산분해효소의 활성을 최소화해야 한다[20]. cfDNA는 생체 외에서 계속 분해되므로, 혈장 분리 직후 cfDNA를 추출하는 것이 권장된다. 혈장을 단기간 보관하기 위해서는 4°C에 3시간 혹은 -20°C에 좀 더 긴 기간 동안 보관할 수 있다[21]. 여러 연구에서 혈장에서 cfDNA의 안정성에 대한 온도와 기간의 영향을 조사하였다[21, 28, 29]. 장기 보관을 위해서는 혈장을 -80°C에 보관하는 것을 권장한다. 장기 보관을 위한 허용 기간은 cfDNA 분석의 목적에 따라 달라질 수 있다[20, 21]. 여러 번의 동결해동 주기를 피하기 위하여 혈장을 검사에 필요한 만큼 소량씩 소분하여 보관하는 것을 권장한다.
4. cfDNA 추출 및 정도관리, 보관
1) cfDNA 추출
cfDNA의 주요한 추출 원리는 스핀 컬럼(spin-column) 기반 방식과 자성 비드(magnetic-bead) 기반 방식이 있다. 스핀 컬럼 기반 방식을 이용하는 키트로는 QIAamp Circulating Nucleic Acid Kit (Qiagen, Hilden, Germany), Quick-cfDNA Serum & Plasma Kit (Zymo Research, Irvine, CA, USA), Plasma/Serum Cell-free Circulating DNA Purification Midi Kit (Norgen Biotek Corp., Thorold, ON, Canada) 등이 있다. 자성 비드 기반 방식을 이용하는 키트로는 QIAamp minElute ccfDNA Mini Kit (Qiagen), Maxwell RSC ccfDNA Plasma Kit (Promega, Madison, WI, USA), MagMAX cell-free DNA Isolation Kit (Thermo Fisher Scientific, Waltham, MA, USA), NextPrep-Mag cfDNA Isolation Kit (Bioo Scientific, Austin, TX, SA), Magnetic Serum/Plasma Circulating DNA Kit (Dxome, Seoul, Korea) 등이 있다. 다양한 cfDNA 추출 키트의 성능을 평가한 연구가 다수 보고되었다[18, 30-34]. 각 검사실에서는 저분자량 DNA의 분리를 위해 수율(yield)과 순도(purity)를 고려한 추출 방법을 선택해야 한다. 또한 추출 장비 및 시약의 성능과 각 검사실에서 필요한 검체 처리량에 따라 수동 혹은 자동화된 작업흐름을 고려할 수 있다.
2) cfDNA 정도관리
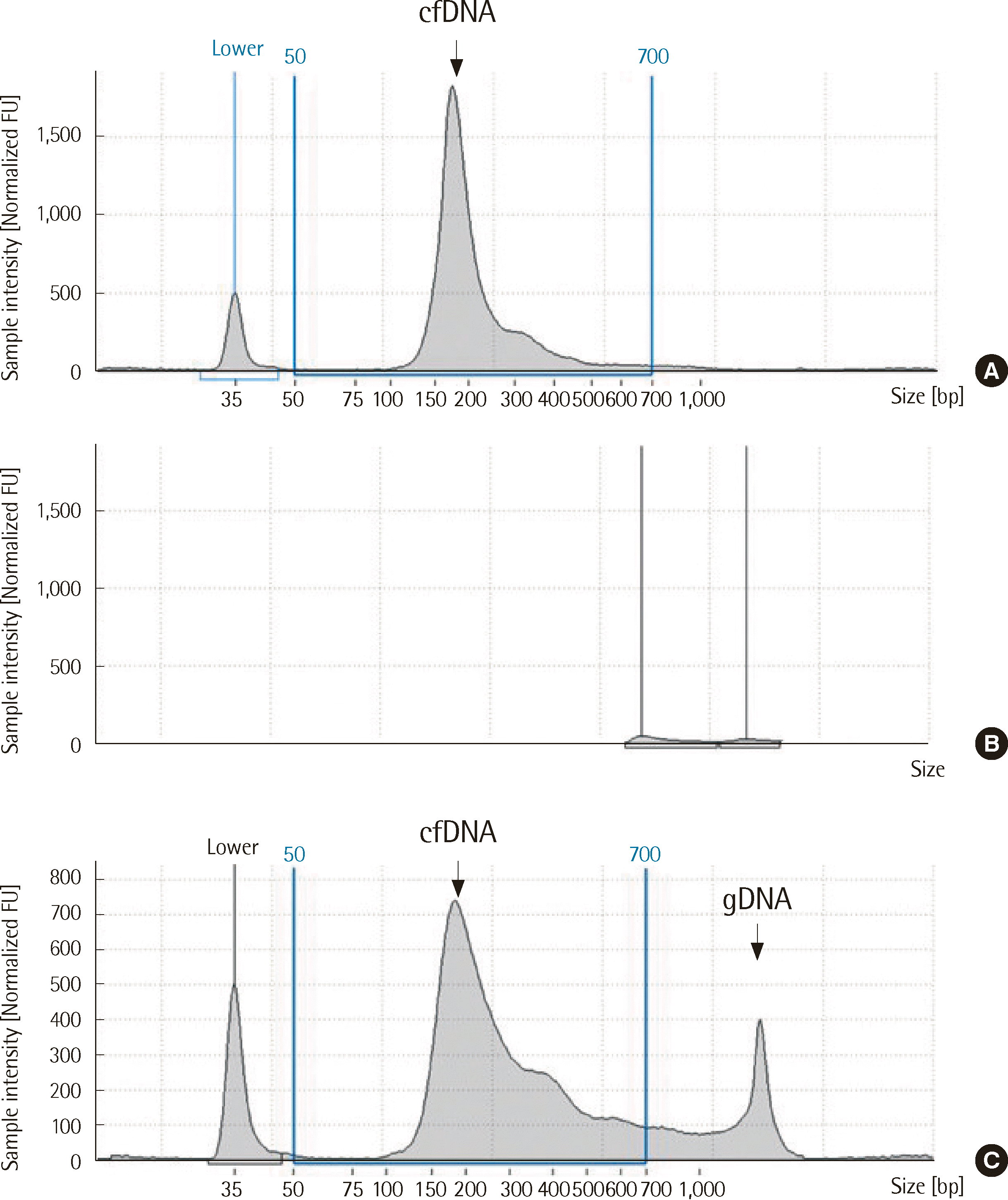
cfDNA 분석을 위해서는 추출한 cfDNA의 양과 분자 크기에 대한 정도관리가 필수적이다. cfDNA의 정량 분석을 위해서는 분광광도법(spectrophotometry; 예, NanoDrop [Thermo Fisher Scientific]) 및 형광분석기(μuorometry; 예, Qubit [Thermo Fisher Scientific] 및 Quantus [Promega]), 실시간중합효소연쇄반응(real-time PCR), 디지털중합효소연쇄반응(digital PCR) 등을 사용할 수 있다[35]. 낮은 농도의 cfDNA를 측정할 때에는 분광광도법보다 형광분석기를 이용한 정량이 더 정확하다[15, 36]. 전기영동 기반 방법(예, Bioanalyzer [Agilent Technologies] 및 TapeStation [Agilent Technologies])은 cfDNA를 정량하고 크기를 측정할 수 있다(Fig. 1).
분석적 측면
1. 대상 유전자
The Cancer Genome Atlas와 International Cancer Genome Consortium은 암 유전체에 대한 대규모 연구를 수행하여 다양한 고형 종양에서의 주요 원인 유전자(key driver genes)를 보고하였다[37, 38]. 이로써 임상 적용에서 치료 결정을 안내하기 위한 차세대염기서열분석법(next-generation sequencing, NGS)의 사용을 포함한 효과적인 치료와 진단 전략이 개발되었다. Memorial Sloan Kettering Cancer Center에서 구축한 OncoKB 데이터베이스(http://oncokb.org)는 진행성 고형암 환자를 치료하기 위한 특정 바이오마커에 대해 Food and Drug Administration (FDA)에서 승인된 약제를 식별하는 중요한 자원이다[39, 40]. 2023년 1월 현재 임상적으로 관련된 FDA 승인에 연계된 유전자 변이 목록은 Table 4에 제시되어 있다.
2. ctDNA 검출을 위한 차세대염기서열분석법(NGS)
ctDNA의 검출을 위한 검사방법은 PCR 기반 검사법과 NGS 기반 검사법으로 분류할 수 있다. PCR 기반 검사법은 일반적으로 특정 암에서 빈번하게 검출되는 특정 체세포 변이(recurrent somatic variants)를 표적으로 하는데, 이러한 분석은 일반적으로 특정 종류의 종양의 약물에 대한 반응과 관련된 변이(예, 비소세포폐암 환자에서의 EGFR L858R, T790M 변이) 확인과 같은 용도로 사용된다. PCR 기반 검사법에는 정량실시간중합효소연쇄반응(quantitative real-time PCR), 디지털 중합효소연쇄반응(digital PCR), BEAMing (beads, emulsions, amplification and magnetics) 검사법이 포함된다. 반면 NGS 기반 검사법은 특정 호발 부위(hotspot)가 아닌 보다 더 넓은 범위의 변이의 검출을 위해 사용되며 단일염기변이(single nucleotide variants, SNV)뿐 아니라 구조적 변이(structural variants)와 복제수 변이(copy number variations)까지 포함한 종양의 광범위한 유전체정보를 제공한다[41, 42]. 다음의 논의는 ctDNA의 검출을 위한NGS 기반 검사법에 초점을 두고자 한다.
평균적으로, 1 mL의 혈장은 2,000 genome equivalents에 해당하는 cfDNA를 포함한다[43]. 일반적으로 매우 낮은 분율로 존재하는 종양 유래 cfDNA (ctDNA의 variant allele frequency [VAF] < 1%인 경우)의 검출은 일반 NGS가 가지는 분석적 민감도의 한계로 인해 어려운데, 일반 NGS 검사법은 변이 검출 한계가 2%에서 5%의 VAF로 제한되기 때문이다[44-46]. 최근, 분자 바코드(molecular barcode) 및 in silico 기법을 이용한 에러(error)제거와 같은 전략을 사용하여 분석적 민감도를 향상시키는 기술이 NGS 검사법에 적용되고 있는데, 이 경우 라이브러리 제작(library preparation) 및 sequencing 과정에서 생기는 에러로부터 VAF 1% 미만의 진양성(true positive)변이를 구분하는 것이 가능해진다.
1) 분자 바코드(Molecular barcoding)
NGS 검사 단계 중, 라이브러리 제작 및 sequencing 과정에서 에러가 발생할 수 있는데, 특히 라이브러리 제작 중 DNA 증폭과정이나 타겟을 선별하기 위해 hybridization capture하는 과정에서도 발생할 수 있다. 또한 sequencing 과정에서 생기는 에러율은 약 0.1%로 예측된다[47]. 이 에러를 최소화시키기 위하여, unique molecular index (UMI)로 불리는 분자 바코드가 사용된다. UMI는 4–14개의 짧은 염기서열로 이루어진 고유의 표지자인데, NGS 라이브러리 제작의 시작단계에서 DNA 단편에 추가되어 동일한 DNA 가닥에서 유래한 서열분석 판독의 식별을 용이하게 한다. Sequencing이 끝난 후, 개별 sequencing read에서 검출된 변이는 에러로 식별되어 해당 sequencing read는 제거되고, 반면 동일한 UMI를 공유하는 모든 sequencing read에서 검출된 변이는 진양성 변이로 식별되어 유지되며 이러한 sequencing read들은 single strand consensus sequence (SSCS)로 그룹화된다[47].
2) In-silico 알고리즘을 통한 에러 교정
분자 바코드 외에도 최근 생물정보학 기술의 발전에 힘입어 in-silico 알고리즘을 기반으로 한 에러 교정이 도입되었고, 해당 기술을 이용하여 NGS 기반 ctDNA 분석의 분석적 민감도가 개선되었다. Pécuchet 등의 최근 연구는 base-position error rates를 소개하였는데, 이는 0.3%까지의 낮은 VAF를 가진 단일핵산변이를 검출하고 삽입/결실 변이의 경우는 0.1%까지의 VAF를 가진 변이를 검출한다[53]. 또한 Newman 등에 의한 다른 연구는 integrated digital error suppression (iDES) enhanced CAPP-Seq기술을 소개하였는데, 해당 기술은 cfDNA 서열분석 데이터에서 탐지된 에러의 in silico 기반 제거와 분자 바코드 기법을 결합하여 0.00025–0.002% 민감도로 암-유래 cfDNA의 검출이 가능하다[54].
3) NGS기반 ctDNA 검사
NGS 기반 ctDNA 분석의 분석적 성능은 과거 연구에서 광범위하게 평가되었다. Deveson 등은 AVENIO ctDNA expanded kit (Roche Diagnostics, Indianapolis, IN, USA), TruSight Tumor 170 (Illumina, San Diego, CA, USA), xGen Non-small Cell Lung Cancer (Integrated DNA Technologies, Coralville, IA, USA), Lung Plasma v4 (Burning Rock Biotech, Guangzhou, China)와 Oncomine Lung cfDNA assay (Thermo Fisher Scientific, USA)를 포함한 5개의 NGS기반 ctDNA 검사의 성능을 평가하였다[55]. 25 ng의 cfDNA로 실험을 진행하였을 때, 각 검사는 서로 다른 unique fragment depth (Burning Rock Biotech and Roche, ~4,700X; Illumina, ~1,200X)를 보였다[56]. 해당 지표는 동일한 분자 바코드를 공유하고 있는 sequence read 들을 취합하여 진양성 변이로 식별되는 변이를 포함한 sequencing read의 depth로, 궁극적으로는 각 검사의 수행 능력에 직접적인 영향을 미친다. Thermo Fisher Scientific의 amplicon 기반 검사는 hybrid capture 검사와 유사한 unique fragment depth를 보였다. Hybrid capture 검사에서는, 0.5% 미만의 낮은 VAF (VAF 범위, 0.1–0.5%)의 변이에서는 검사들 간의 분석적 민감도 차이(분석적 민감도의 범위, 0.39–0.83)가 관찰되었다. Integrated DNA Technologies와 Burning Rock Biotech 검사는 VAF 범위가 0.3–0.5%인 변이에서 분석적 민감도 0.90 이상을 보여 가장 민감하였다.
Koessler 등에 의한 다른 연구에서는 Oncomine Lung cfDNA assay (ThermoFisher), Avenio ctDNA expanded kit (Roche)와 QIAseq human lung cancer panel (Qiagen) 포함 3가지 서로 다른 NGS 기반 ctDNA 분석을 비교하였는데, 해당 연구에 따르면 VAF 최소 1%인 변이까지는 안정적이게 검출할 수 있었으나, VAF 0.1%까지 낮아질 경우, 안정적인 검출은 쉽지 않음을 보고하였다[57]. QIAseq 플랫폼은 0.1% VAF의 2개 EGFR 유전자의 삽입/결실 변이 탐지에 실패하였고 0.5% VAF의 샘플에서는 VAF가 낮게 정량되었다.
한국에서는 몇 가지 NGS 기반 ctDNA 검사가 출시되어, 이용가능하다. 몇 임상 검사실은 국내 또는 수입 시약을 사용하여 ctDNA NGS 서비스를 제공하고 있다. 이 중에는 Dxome사의 DxLiquid Pan100 및 DxLiquid TMB500, IMBDdx사의 AlphaLiquid 100, Illumin사의 TruSight Oncology 500 ctDNA, Thermo Fisher Scientific사의 Oncomine Pan-Cancer Cell-Free Assay 등이 있다. ctDNA 검사 서비스의 경우, 해외 공급업체인 Guardant Health, Inc.를 통한 Guardant360 NGS assay 및 Foundation Medicine을 통한 FoundationOne Liquid CDx 검사 서비스 이용이 가능하다. 이 경우 검체는 Clinical Laboratory Improvement Amendment 인증을 획득한 미국 내 검사실로 보내지며 결과보고서는 한국으로 송부된다. 다만, 해당 서비스는 국내 건강보험 적용 대상은 아니다.
추가로, NGS 라이브러리 제작, 염기서열분석, 생물정보학 분석이 각각 적절하게 수행되는지 모니터링하기 위하여 검사의 질관리(quality control, QC) 절차를 확립하는 것이 필요하다. NGS기반 ctDNA 분석 모니터링을 위한 유용한 QC 지표에는, cfDNA quality와 quantity, library qualification과 quantification, base call quality scores, cluster density, sequenced read pairs의 개수, GC bias, alignment rate, transition/transversion ratio, mapping quality, duplication rate, strand bias, sequencing depth, unique depth, on-target rate, coverage uniformity 등이 있다[58].
3. 검사의 평가
현재 임상 검사실에서 사용되는 대부분의 NGS기반 ctDNA 검사는 laboratory-developed tests (LDTs)이며, 이들의 성능은 이미 기확립된 임상적 평가 절차 및 표준을 통한 검증이 필요하다. 최근 액체생검(liquid biopsy)의 개발, 평가, 임상적 사용 전반에 걸친 기존의 어려움을 돌파하고자 Blood Profiling Atlas in Cancer (BloodPAC) working group이 조직되었다[59]. 이 단체는 NGS 기반 ctDNA 분석에 특히 중점을 둔 분석적 성능 평가에 대한 권장 프로토콜을 개발하였다. BloodPAC working group의 경우 혈액에서 추출된 cfDNA가 매우 극소량이라는 점, 그리고 그중 종양 유래 cfDNA의 분율이 매우 낮다는 점에서 비롯되는 어려움들을 고찰하고 있는데, 여기서의 어려움이란 ctDNA 검출을 위한 민감한 검사의 필요성, 낮은 VAF로 존재하는 ctDNA에 대한 위음성 결과의 가능성, 그리고 충분한 양의 ctDNA 확보를 위한 contrived sample의 필요성을 포함한다. BloodPAC working group은 NGS 기반 ctDNA 분석를 위한 분석 검증 프로토콜을 확립하였는데, 이는 limit of detection (LOD), limit of quantification (LOQ), analytical accuracy, linearity, precision, 그리고 interference를 포함한다[60].
현재 국내에는 NGS 기반 ctDNA 검사 평가에 대한 지침이 부족한 실정이다. 그 대안으로, 국내 식품의약품안전청은 NGS 검사 전반에 초점을 맞추어, NGS 검사를 도입할 때 임상검사실에서 수행해야 하는 분석적 성능 및 임상적 성능 평가에 대한 자세한 프로토콜을 제공한다. 분석적 성능 평가에 포함되는 항목은 검출한계(limit of detection), 측정 범위, 판정 기준치 등을 포함하는 분석적 민감도, 분석적 특이도, 정밀도, 정확도 및 교차반응이 있다. 임상적 성능 평가는 임상적 민감도와 임상적 특이도를 평가하는데, 각 항목에 대해 해당 프로토콜은 일반적인 설명, 시험 물질, 시험 방법, 결과 제시 방법을 기술하고 있다[61]. 이외에도, 진단검사의학재단과 한국유전자검사평가원은 검사실 인증평가를 통해 임상 검사실의 질관리를 향상하고 신뢰할 수 있는 검사결과를 얻을 수 있도록 노력하고 있다. 이들 기관은 특히 LDT 검사의 성능을 평가하는 검증과정 및 질 관리 방법을 정의하는 문서화된 지침을 제공한다. 각 지침은 각각의 임상검사실이 LDT 검사의 성능 평가를 위한 구체적인 지침서를 갖추도록 권장하고 있다. LDT 검사의 성능 평가는 분석적 성능 평가(정확도, 상관성, 정밀도, 직선성, 보고범위, 검출한계, 정량한계 및 간섭) 및 임상적 성능 평가(임상적 민감도 및 특이도, 양성 및 음성 예측도)를 포함해야 한다[62, 63].
해석 및 보고
1. 변이 해석
변이 해석은 생물학적 해석(biological interpretation)과 임상적 해석(clinical interpretation)의 두 가지 과정으로 분류된다. 생물학적 해석은 변이의 종양원성(oncogenicity)을 결정하는 과정이며, 2015년 발표된 미국의학유전학회와 분자병리학회(ACMG/AMP)의 가이드라인[64]이 생식세포 변이(germline variants)에 대한 생물학적 해석의 표준으로 간주된다. 2015 ACMG/AMP 가이드라인은 인구집단 자료, 예측 결과, 기능 연구, 분리(segregation) 자료, de novo 여부, 대립유전자 근거(cis/trans) 등의 기준에 따라 변이를 5가지 범주(pathogenic, likely pathogenic, variant of uncertain significance, likely benign, benign)로 분류한다.
추가적으로 Clinical Genome Resource (ClinGen)의 sequence variant interpretation (SVI) working group에서는 특정 유전자 및 질병의 고유한 특성을 잘 반영할 수 있도록 유전자 및 질병 특이적인 가이드라인을 발표하고 있다[65]. 그러나 이러한 가이드라인은 생식세포 변이의 병원성(pathogenicity)을 판단하는 데 적합하였다. 2022년 5월, 체세포 변이(somatic variants)의 종양원성을 결정하기 위한 가이드라인이 ClinGen, Cancer Genomics Consortium (CGC), Variant Interpretation for Cancer Consortium (VICC)에 의해 발표되었다[66]. ClinGen/CGC/VICC 가이드라인은 인구집단 자료, 기능 연구, 변이 종류, 핫스팟, 예측 결과 등의 기준에 따라 변이를 5가지 범주(oncogenic, likely oncogenic, variant of uncertain significance, likely benign, benign)로 분류한다.
임상적 해석은 변이의 actionability를 결정하는 과정이며, 2017년 발표된 AMP/미국임상종양학회(American Society of Clinical Oncology, ASCO)/미국병리학회(College of American Pathologists, CAP) 가이드라인이 체세포 변이의 임상적 해석을 위해 널리 사용되고 있다[67]. 2017 AMP/ASCO/CAP 가이드라인은 변이의 임상적 영향을 결정하기 위해 4단계의 근거(level A, B, C, D)를 사용한다. 이러한 근거를 바탕으로 변이는 진단, 예후 및 치료와 관련된 임상적 중요도에 따라 4개의 tier (I, II, III, IV)로 분류된다. Tier I은 강한 임상적 중요성을 갖는 변이(level A 및 B 근거), Tier II는 잠재적 임상적 중요성을 갖는 변이(level C 또는 D 근거), Tier III는 임상적 중요성이 불확실한 변이, Tier IV는 benign 또는 likely benign 변이이다. 이 외에도 European Society for Medical Oncology Scale of Clinical Actionability for molecular Targets (ESCAT) 가이드라인에서는 환자 관리에 미치는 영향에 따라 변이를 6개의 tier (I, II, III, IV, V, X)로 분류한다[68].
Precision Oncology Knowledge Base (OncoKB)는 체세포 변이의 생물학적 및 임상적 해석에 대한 주석을 제공한다[40]. 임상적 해석을 위해서 OncoKB는 OncoKB Levels of Evidence system을 사용하여 변이를 6가지 수준으로 분류한다(민감도에 대하여 Level 1-4, 저항성에 대하여 Level R1 및 R2). OncoKB의 분류는 2017 AMP/ASCO/CAP 가이드라인의 Tier와 맞춰볼 수 있는데 이는 다음과 같다: OncoKB level 1, 2 및 R1은 Tier 1A, OncoKB level 3A는 Tier 1B, OncoKB level 3B는 Tier IIB, OncoKB level 4 및 R2는 Tier 2D에 해당한다. 또한 생물학적 해석을 위해서 변이는 종양원성에 따라 oncogenic, likely oncogenic, likely neutral, and inconclusive로 분류되며, ClinGen/CGC/VICC 가이드라인과의 일치 여부는 아직 확립되지 않았다. OncoKB에서 제공하는 변이의 분류는 OncoKB 웹사이트 및 cBioPortal for Cancer Genomics (http://www.cbioportal.org)를 통해 확인할 수 있다. 이 외에 변이 해석을 위해 참고할 수 있는 다양한 데이터베이스들이 있으며, 이 중 Cancer Genome Interpreter Cancer Biomarkers Database (CGI), Clinical Interpretation of Variants in Cancer (CIViC), Jackson Laboratory Clinical Knowledgebase (JAX-CKB), MolecularMatch (MMatch), OncoKB, Precision Medicine Knowledgebase (PMKB) 등 6개 지식베이스를 통합한 meta-knowledgebase로 VICC가 있다[69].
체세포 변이에 대한 해석은 대부분 임상적 해석에 의존해 왔다. 실제로 고형암 NGS에 대한 CAP 숙련도 검사에 참여한 152개 기관을 대상으로 조사한 결과[70], 84.9% (129/152)가 체세포 변이 해석에 2017년 AMP/ASCO/CAP 가이드라인을 사용한다고 답변했다. 이 중 68.2% (88/129)는 오직 2017 AMP/ASCO/CAP 가이드라인만을 사용하였으며, 나머지 31.8% (41/129)는 2017 AMP/ASCO/CAP 가이드라인을 다른 가이드라인과 함께 사용한다고 답변했다. 함께 사용한 가이드라인의 대부분은 2015 ACMG/AMP 가이드라인이었다. 그러나 체세포 변이를 종합적으로 이해하려면 체세포 변이의 생물학적 측면과 임상적 측면을 모두 고려하는 것이 중요하다. 따라서 임상적 해석과 생물학적 해석을 모두 사용하여 체세포 변이를 평가하는 것이 고려될 수 있다.
정밀의료를 구현할 때 다학제 전문가와 함께 유전자 결과를 바탕으로 잠재적 치료 옵션을 논의하는 것이 중요하며, 이는 환자 관리를 위한 임상적 의사결정 과정에 도움이 된다. 따라서 각 기관은 유전학, 종양학, 병리학, 유전상담학, 생물정보학 등 다학제 전문가로 구성된 분자종양위원회(molecular tumor board, MTB)를 구성할 수 있다. MTB를 통해 개별 환자 사례를 종합적으로 평가하고 표적 치료법 사용을 최적화할 수 있으며, 따라서 환자는 가장 적합한 치료법을 받거나 새로운 치료법에 대한 임상시험에 참여할 수 있다. Larson 등의 체계적 고찰에 따르면[71], MTB를 통해 권장된 치료를 받은 환자는 전통적인 치료를 받은 환자에 비해 임상 결과가 개선되었다.
2. 변이 해석 시 고려사항
1) 낮은 대립유전자 빈도(variant allele frequency, VAF)
VAF가 낮은 변이가 발견된 경우 실제 변이인지 위양성인지 구별하기 어려울 수 있으므로 VAF가 낮은 변이의 의미를 해석하는 것은 어려운 일이다. 종양세포로부터 유래하지 않은 cfDNA는 ctDNA를 희석시키기 떄문에 ctDNA 검사에서의 낮은 VAF를 초래하며, 이는 낮은 종양 부하, subclonal 변이의 존재 또는 뇌 전이와 같이 ctDNA의 양이 낮은 경우에 특히 문제가 된다. 낮은 VAF 변이의 임상적 중요성은 불확실하지만, 일부 연구에 따르면 driver genes에서 발견되는 변이는 낮은 VAF에서도 표적 치료에 잘 반응하는 것으로 나타났다[72, 73]. 따라서 실제 변이와 위양성을 정확하게 구분하는 것이 중요하다. 검출 한계(limit of detection, LOD) 미만의 변이는 추가 검증이 수행되지 않는 한 보고해서는 안 되며, sequencing 깊이를 늘리는 것이 진양성 변이를 식별하고 위양성 가능성을 줄이는 데 도움이 될 수 있다.
2) Clonal hematopoiesis of indeterminate potential (CHIP)
CHIP는 조혈모세포에 체세포 변이가 축적되어 세포의 클론 확장을 초래하는 과정을 말한다. 이는 정상적인 노화 과정에 해당하며 65세 이상 성인의 약 10%에서 보고되고 있다[74]. cfDNA는 대부분 조혈모세포에서 유래하기 때문에 CHIP은 ctDNA 결과를 해석하는 데 중요한 교란 요인으로 작용한다. 특히 DNMT3A, TET2, ASXL1과 같이 일반적으로 CHIP에 관여하는 것으로 알려진 유전자에서 확인된 낮은 VAF 변이의 경우 더욱 문제가 된다[74]. 또한, 일반적으로 고형암과 관련된 것으로 알려진 KRAS, GNAS, NRAS, PIK3CA 등의 유전자에서도 CHIP이 보고되고 있어[75], 해석의 복잡성을 더한다. ctDNA 유래 변이와 CHIP 관련 변이를 구별하기 위해 paired peripheral blood mononuclear cell (PBMC) sequencing을 사용할 수 있다. 또한 ctDNA는 비종양세포에서 유래된 cfDNA보다 길이가 짧기 때문에 생물정보학을 통해 단편 크기를 선택하는 것이 도움이 될 수 있으며 PBMC sequencing에 대한 효과적인 대안이 될 수 있다[76].
3) 우연히 발견된 생식세포 변이
ctDNA 검사에서는 체세포 변이뿐만 아니라 생식세포 변이도 검출될 수 있다. 특히 분석하는 유전자 수가 증가함에 따라 생식세포 변이 검출 가능성도 점점 높아지고 있다. 생식세포 변이는 ctDNA 결과에서 우연히 검출될 수 있으며, 환자에게 이러한 가능성을 알려야 한다는 점에 유의해야 한다. 일반적으로 체세포 변이로 판단하는 데 사용되는 기준에는 VAF 50% 미만, 암에서 임상적 중요성이 알려진 핫스팟 변이, 인구집단 데이터베이스에서 일반적으로 관찰되지 않는 변이 등이 있다[1]. 반면, 약 50% 및 100%의 VAF는 일반적으로 각각 이형접합 및 동형접합 생식세포 변이를 시사한다. 그러나 생식세포 변이의 VAF가 예상과 다를 수 있으므로 주의가 필요하다[77].
ctDNA 결과에서 생식세포 병원성 변이가 의심되는 경우, 확진을 위해 정상 조직을 이용한 생식세포 검사를 시행하고 환자에게 유전상담을 제공하는 것을 고려할 수 있다. 또한 각 검사실은 생식세포 변이 해석 및 보고에 대한 정책을 수립해야 한다. 우연히 발견된 생식세포 변이는 2015 ACMG/AMP 지침에 따라 해석해야 하며, secondary findings 보고에 대한 ACMG 권고사항에 따라 보고해야 한다[78]. CHIP 관련 변이를 필터링하기 위해 이미 PBMC로 정상 조직 검사를 수행하고 있는 경우 생식세포 변이도 확인할 수 있지만, 만약 분석 과정에서 생식세포 변이를 제거하는 것이 실험실의 전략이라면[79] 분석 중에 생식세포 변이가 제거되었음을 결과보고서에 명시해야 한다. 확진을 위한 생식세포 검사를 실시하지 않는 경우, 환자에게 생식세포와 체세포 변이를 구별하지 못할 수 있음을 안내해야 한다. 발병시기, 양측성, 가족력과 같은 임상 정보들이 암 소인 증후군과 관련된 유전자에서 발견된 변이를 해석하는 데 도움이 될 수 있으며[8], 문헌 검토 및 질병 데이터베이스도 생식세포 변이를 식별하는 데 도움이 된다.
4) 조직 검사 결과와의 불일치
ctDNA 결과는 조직으로 수행한 결과와 차이가 있을 수 있다. ctDNA에서 변이가 검출되지 않은 경우 진음성일 수도 있지만 위음성의 가능성을 배제할 수 없다. 위음성은 혈장 내 존재하는 종양 DNA의 양이 변이를 검출하기에 충분하지 않을 정도로 적은 경우에 발생할 수 있다. 특히 중추신경계 암이나 뇌 전이의 경우 혈뇌장벽에 의해 종양 DNA의 방출이 제한되므로 위음성을 일으킬 가능성이 있으며, 이러한 경우 뇌척수액을 이용한 ctDNA 검사가 유용할 수 있다. 결과 보고 시에는 이러한 한계를 환자에게 안내해야 하며 “negative” 대신 “not detected”, “undetected” 또는 “uninformative”와 같은 용어를 사용하는 것이 바람직하다[1]. 반면에 조직에서 검출되지 않은 변이가 ctDNA에서만 검출될 수도 있는데 이는 조직 검사에서 종양 이질성(tumor heterogeneity)이 잘 반영되지 않기 때문일 수 있다[80]. 따라서 치료의 표적이 되는 변이를 조직에서 검출할 수 없을 때 ctDNA가 추가적인 치료 옵션을 제공할 가능성이 있다.
3. 결과보고서 작성
ctDNA 결과보고서는 임상적 의사결정에 필수적인 정보를 포함해야 하며, 2페이지 이내로 간결하게 작성하는 것이 이상적이다. 기본적으로 환자 및 검체 정보가 포함되어야 하며, 환자 정보에는 환자의 이름, 성별, 나이, 종양 유형 및 조직학적 결과가 포함된다. 검체 정보에는 검체번호, 검체 유형, 및 검체채취 날짜가 포함된다.
발견된 변이는 Human Genome Variation Society (HGVS)의 표준 명명법(http://varnomen.hgvs.org/)에 따라 coding DNA 및 단백질 수준에서 기술해야 한다. HUGO Gene Nomenclature Committee (https://www.genenames.org/)에서 승인된 유전자명과 Matched Annotation from NCBI and EMBL-EBI (MANE) Select (https://www.ncbi.nlm.nih.gov/refseq/MANE)에 따른 표준염기서열을 사용하여 변이를 기술해야 한다. 또한 변이에 대한 명확한 의사소통과 이해를 위해 표준 명명법 외에 일반적으로 사용되는 명명법을 사용하여 변이를 기술할 수 있다[67].
돌연변이 부하(mutant burden)는 VAF를 사용하여 보고할 수 있다. 이전 연구에 의하면 종양 크기는 ctDNA의 VAF와 관련이 있다[81, 82]. 따라서 VAF는 종양 부하를 추정하는 데 사용할 수 있다. 또한 동일한 검체에서 변이들의 VAF를 비교함으로써 종양 이질성을 반영하는 subclone 변이를 식별할 수 있다[83]. 그러나 백혈구 DNA의 양은 분석 전 단계로 인해 검체마다 차이가 있을 수 있다. 따라서 VAF를 신중하게 해석하는 것이 중요하며, 이러한 경우 돌연변이 부하를 copies/ml of plasma로 보고하는 것이 도움이 될 수 있다[84, 85].
변이에 대한 임상적 해석은 적절한 치료 전략 선택에 대한 정보를 제공하기 때문에 보고서에서 가장 중요한 부분이다. 변이는 2017 AMP/ASCO/CAP 가이드라인에 따라 4가지 tier로 분류해야 한다. Tier 1부터 3까지는 보고서에 포함해야 하며 tier 4는 임상적 중요성이 없는 benign 또는 likely benign 변이이므로 포함하지 않아야 한다[67]. 가능한 경우, 종양원성도 보고서에 포함할 수 있다. 동일한 변이라도 종양 유형에 따라 임상 결과가 다를 수 있으므로 임상적 해석은 환자의 종양 유형에 따라 수행되어야 한다. 치료의 임상 결과는 유전 정보 외에도 여러 요인에 따라 달라지므로 유전 정보에 기반한 약물 추천은 구체적이어서는 안 되며, 결과보고서에는 변이와 약물 간의 일반적인 연관성을 기술하는 것이 좋다[1]. 예를 들면, 특정 약물을 사용할 것을 추천하지 말고, 대신 변이와 약물의 효능과의 관계를 설명하는 것이 적절하다. 변이와 약물의 효능과의 관계를 설명할 때는 환자의 종양 유형과 관련이 있어야 하며 근거에 대한 참고 문헌을 인용해야 한다[67]. 각 검사실은 최신 정보를 정기적으로 업데이트해서 환자에게 적절한 치료를 놓치는 일이 없도록 해야 한다.
미세부수체 불안정성(microsatellite instability, MSI)과 종양변이 부담(tumor mutational burden, TMB)은 면역관문억제제에 대한 종양 불문 바이오마커(tumor-agnostic biomarker)이다. 여러 연구에 따르면 ctDNA로 추정된 blood MSI 및 blood TMB (bMSI 및 bTMB)는 tissue MSI 및 tissue TMB (tMSI 및 tTMB)와 강한 상관관계를 보인다고 보고된 바 있다[86-89]. 하지만 현재까지 pembrolizumab의 동반 진단으로 FDA 승인을 받은 것은 Foundation Medicine사의 FoundationOne CDx를 이용한 tMSI 및 tTMB가 유일하다[69, 90]. 따라서 ctDNA를 통한 bMSI 및 bTMB의 활용을 위해서 bMSI 및 bTMB의 임상적 효능 및 최적의 cutoff에 대한 추가 연구가 필요하다.
결과보고서에는 검사 방법에 대한 세부 정보가 포함되어야 한다. 여기에는 검사 방법, 검사범위에 포함되는 유전자 및 영역, 참조 유전체, 분석 성능, 중요 품질 지표, 검사의 한계가 포함된다. 전체 유전자가 검사 범위가 아닌 경우에는 엑손 또는 핫스팟과 같이 실제로 검사된 영역을 표시해야 한다. 또한 생물학적 또는 기술적 이유로 인해 sequencing의 최소 깊이를 충족하지 못한 영역도 표시해야 한다. 검사의 성공 여부를 판단하기 위해 LOD 또는 최소 sequencing 깊이와 같은 분석 성능과 input DNA 양 및 sequencing 깊이와 같은 중요 품질 지표를 포함할 수 있다. 변이가 검출되지 않았을 경우 낮은 민감도로 인한 위음성 가능성을 한계로 기술해야 한다. 특히, 비소세포폐암의 EGFR과 같이 암 특이적인 actionable 유전자에서 Tier 1 변이가 발견되지 않은 경우에는 변이가 발견되지 않았다는 사실을 언급하고 종양 조직을 이용한 검사를 권고할 수 있다[67, 91]. 따라서 각 기관에서는 종양에 따른 actionable 유전자 목록을 보유하고 있어야 한다. 결과 해석 및 보고에 대한 권장 사항은 Table 5에 요약되어 있다.
 XML Download
XML Download