

 PDF
PDF Citation
Citation Print
Print



Introduction
Stroke is a type of cerebrovascular disease which commonly occurs in adults and elderly. It remains the worldwide third majority cause of disability and death [1]. The incident of stroke cases infinitely increased to 70% from 1990 to 2019, and the mortality rate escalated to 40% [2]. More than 87% of strokes are presented as ischemia [3]. Cerebral ischemia or ischemic stroke is the insufficient blood supply and nutrient to the brain caused by cerebral artery occlusion leading to brain dysfunction sand damage [4]. The common forms of occlusion resulting in blood flow obstruction to the brain are thrombosis, the ruptured plaque from cerebral stenosis or atherosclerosis, and embolism, the clot formed in vessel and heart [5]. The severity of brain damage depends on occlusion time. Recombinant tissue-type plasminogen activator is recommended for intravenous injection within 4.5 hours after stroke symptoms occurring. The golden hour for ischemic stroke treatment is less than 1 hour [6, 7]. Long-term cerebral artery occlusion without re-perfusion leads to permanent brain injury. There are many homeostatic mechanisms occurring after cerebral vascular obstruction. Excessive glutamate releasing and calcium toxicity is the common stimulator arising in acute stage of ischemia, which can lead to various brain damage pathways [8]. Moreover, it involves the autophagic impairment and neuronal death stimulation during acute phase [9]. This article reviews the underlying mechanism of calcium toxicity related to autophagy up-regulation in acute stage of permanent cerebral ischemia.
Review
Cerebral ischemia in acute stage
Anoxic depolarization and glutamate excitotoxicity
Brain is the high energy consumption organ which mainly synthesizes more than 20% of total adenosine triphosphate (ATP) to retain its functions [10]. Neurons drive the neuronal signaling by conveying the electrochemical ions via ATPase ion pumps on cell membrane, called action potential [11, 12]. Neurotransmitters play a vital role in neuronal conducting of neuron to neuron both excitatory and inhibitory signaling [13, 14].
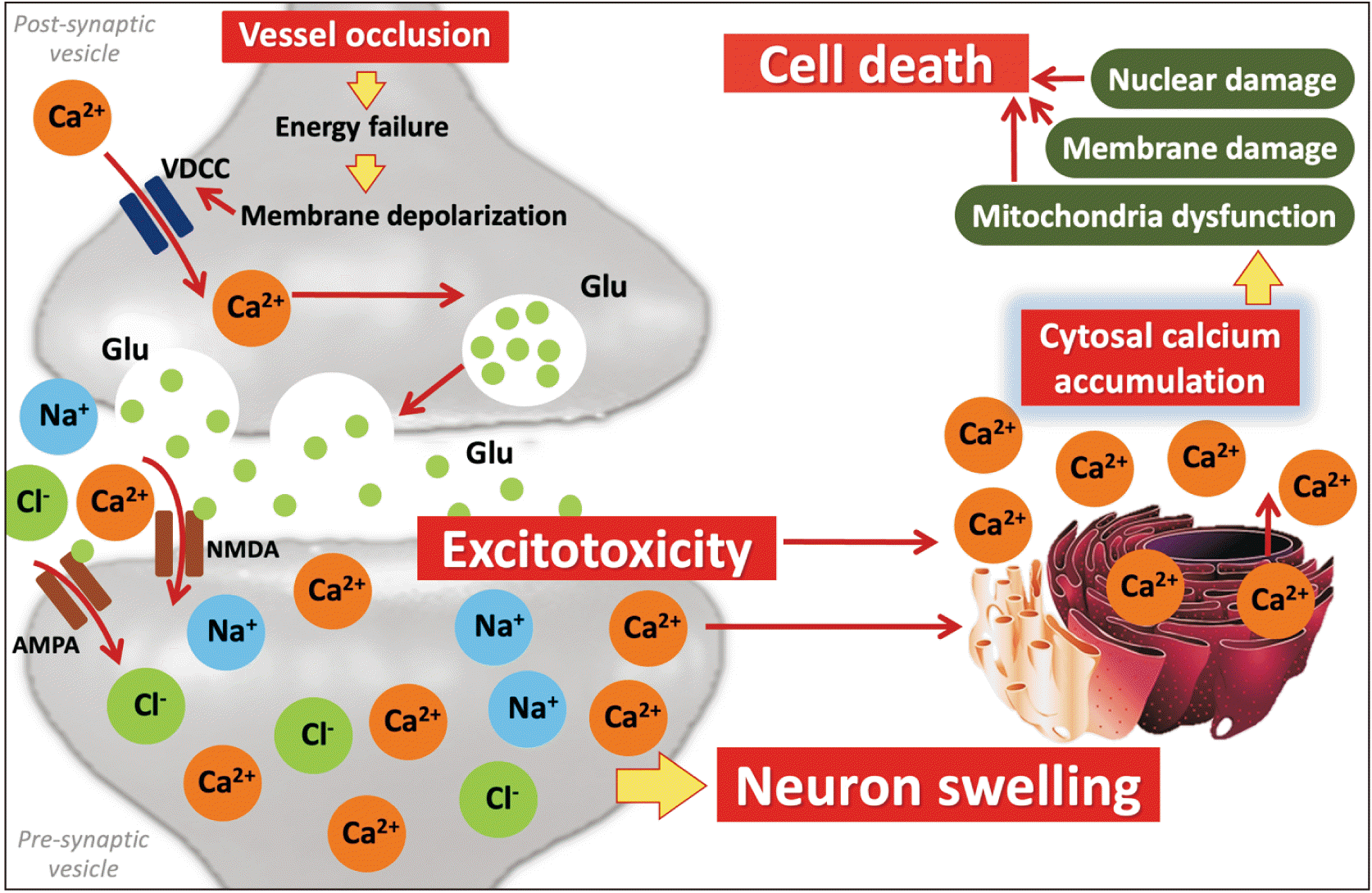
After the cerebral artery occlusion, decreased cerebral blood flow to the brain leads to reduced oxygen and glucose consumption because of depleted ATP produced from mitochondria in neuron and glial cell [15, 16]. These result in ATPase ion pumps failure, known as anoxic depolarization including Na+/K+-ATPase (NKA), Na+/Ca2+-ATPase pumps (NCX) and Ca2+ ATPase channels. Generally, NKA pumps 2 ions of K+ into neurons to evoke action potential and offers energy to NCX pumps which control the concentration of Ca2+, Na+ and K+ between extra- and intracellular space of neuronal cells [17-19]. Impaired ATPase ion pumps affect the flowing of ions into the cell and results in neuronal swelling [20]. Likewise, depleted ATP provokes voltage-gated calcium channel (VGCC) to maintain function resulting in intracellular Ca2+ overload [21] Moreover, these engenders Ca2+ influx into presynaptic area leading to excessive glutamate releasing to synaptic space with glutamate re-uptake failure [22].
Glutamate is an excitatory neurotransmitter related to learning and memory, which is released from presynaptic terminals to stimulate other neuronal cells [23]. Neurons and astrocytes are the two major cells associated with glutamate metabolism [24]. Glutamate releasing activates ionotropic glutamate receptors and ligand-gated ion channels on postsynaptic membranes which rapidly responses to allow ions influx and stimulate various downstream cascades [25]. Under ischemic conditions, excessive glutamate releasing, or glutamate excitotoxicity is the main point which triggers neuronal dysfunction in acute stage [26]. Immoderate glutamate receptor activation promotes Ca2+ transferring into the cell, which can provoke a various cellular signaling resulting to neuronal death [27]. In addition, the dysfunction of ion exchange across neuronal membrane impels exorbitant Na+ influx causing hyperosmotic movement and cell swelling, respectively (Fig. 1) [28, 29].
NMDAr activation and calcium toxicity
N-methyl-D-aspartate receptor (NMDAr) is a type of ligand-gated ion channel which plays an important role in early ischemia. Over activation of NMDAr by unnecessary glutamate releasing affects to intracellular Ca2+ accumulation resulting to calcium toxicity, which can promote many signaling cascade associated with neuronal dysfunction and death [30, 31]. Excessive NMDAr stimulation is the major cause of neurotoxicity in acute ischemia. Abnormal Ca2-dependent enzyme activation led to cell death signaling. NMDAr consists of 2 main heterotetrametric forms including GluN1 and GluN2 subunits. These subunits have an important function in neuronal survival and neuronal death. GluN2 subunit is separated into 2 subtypes; GluN2A and GlutN2B. GluN2A is greatly expressed at synapse site of neuron which plays a key role in neuronal survival [32, 33]. The activated GluN2A can activate phosphoinositide 3-kinase (PI3K) by binding to Ca2+ and calmodulin which phosphorylates protein kinase B (Akt), respectively. Phosphorylated Akt generally inhibits pro-apoptotic factors [21, 34]. Moreover, the activated NMDAr stimulates mitogen-activated protein kinases/extracellular signal-regulated kinases (ERK) pathway, the extracellular signaling regulated protein kinase and Ca2+/calmodulin-dependent protein kinase, the calcium signaling, that can provoke cAMP response element-binding protein (CREB) by phosphorylation to motivate anti-apoptotic proteins [35].
Under ischemic condition, excessive glutamate releasing extremely stimulates NMDAr, especially GluN2B subtype, which promote pro-apoptotic cell death signaling cascade [36]. GluN2B is fully located at an extra-synaptic site. Over-synaptic NMDAr can trigger GluN2B subtype to dephosphorylate and inhibit ERK and CREB signaling pathway resulting to pro-apoptotic activation [35, 37]. Furthermore, GluN2B also provokes postsynaptic density protein 95 (PSD95) protein which downstream stimulates neuronal nitric oxide synthase (nNOS) by binding to the N-terminus, called GluN2B/PSD95/nNOS complex, leading to nitric oxide (NO) production [38, 39]. NO interacts with superoxide radical molecules and critically forms reactive nitrogen species (RNS) that are associated with protein oxidation, lipid peroxidation and DNA fragmentation [40].
Death associated protein kinase 1 (DAPK1), the mediator induced programmed cell death, is disinhibited from autophosphorylation suppressing by high level of intracellular Ca2+ activated calmodulin (CaM) and leads to pro-apoptotic activation [41]. Likewise, DAPK1 can directly interact with the C-terminal region of GluN2B tail to escalate the stimulation of GluN2B subtype. GluN2B/DAPK1 binding complex encourages more severity of neuronal damage [42]. Furthermore, activated DAPK1 can interact with programmed cell death 6 (PDCD6), tumor protein 53 and protein kinase D to promote both necrotic and apoptotic neuronal cell death [42-44].
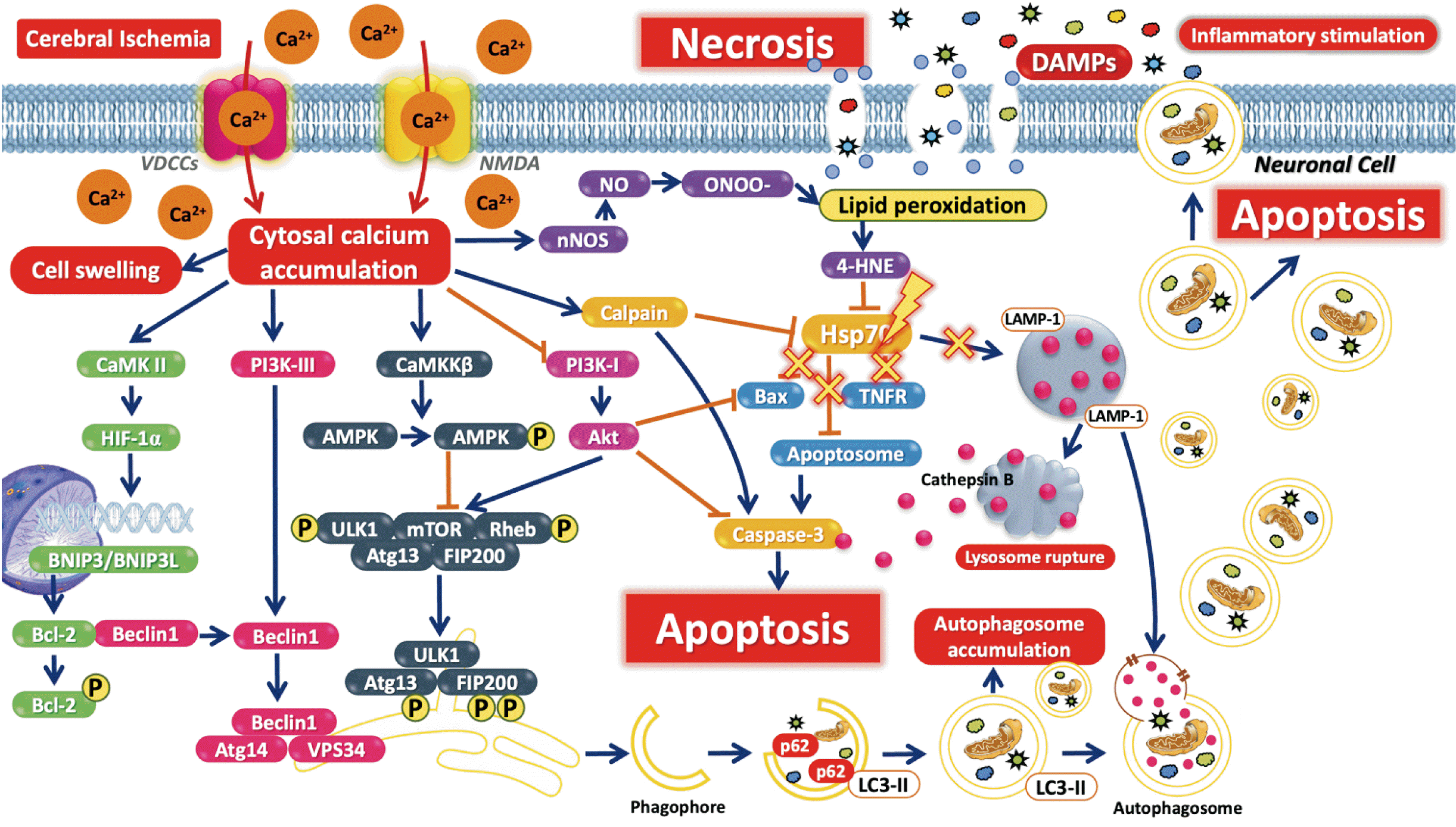
Over NMDAr activation vastly fluxes Ca2+ into the neuron [45]. There is reported Ca2+ quickly influxes into the neuronal cell via NMDAr in the early stage of ischemia [46]. High Ca2+ concentration accumulated in cytoplasm can trigger many downstream cascades of neuronal death signaling [8]. Calpain (CAPN) is a Ca2+-dependent cysteines protease which is greatly found in central nervous system [47]. In the brain, CAPN acts as Ca2+-dependent neuronal function controlling. CAPN is separated into 2 major isoforms which are CAPN1 or μ-calpain and CAPN2 or m-calpain [48]. In ischemic brain, CAPN1 is activated by excessive Ca2+ accumulation. Activated CAPN1 play a key role in neuronal death. CAPN1 is the protease enzyme that hydrolyzes various cytoskeleton proteins causing neuronal dysfunction and apoptotic stimulation [49, 50]. Additionally, overactivated NMDAr downstream stimulates CAPN1 in cytosol which can cleave NCX channels at neuronal membrane resulting in promoting NCX channels dysfunction and poorly control Ca2+ influx [51]. Moreover, CAPN1 also affects to metabotropic glutamate receptor 1 protein at cell membrane that normally motivates brain function and acts as neuroprotective by interacting with nuclear phosphoinositide-3-kinase enhancer to promote PI3K/Akt signaling pathway (Fig. 2) [52].
Autophagic cell death
Autophagy is a self-digestive program for cellular homeostatic maintaining and cell survival by degrading misfold proteins and damaged organelles to produce energy [53]. Macro-autophagy is the general form of autophagic process normally found in eukaryotic cells [54]. The common characteristics are autophagosome forming and fuses with lysosome to degrade the deformed proteins and dysfunctional organelles [55]. The major key of autophagic initiation is mammalian target of rapamycin (mTOR) which is phosphatidylinositol 3-kinase-related kinase family and controls cellular physiology, protein synthesis and autophagy [56]. mTOR regulates autophagy by phosphorylation of Atg1/ULK1 complex to form autophagopore with beclin-1 [57]. Beclin-1 is the autophagy regulator conjugated with PI3K class III to form beclin-1/Vsp34/Atg14/PI3K-III complex and stimulate autophagopore formation [58]. Microtubule-associated proteins 1A/1B light chain 3A (LC3) are the main protein of autophagosome establishing. LC3-II plays an important role in double membrane elongation to form vesicles which engulf the damaged organelles and proteins, known as autophagosome [59]. Autophagosome is fused with lysosome called autophagolysosome to degrade the products by acid hydrolase within lysosome and obtain the final products as free fatty acids and amino acids to generate energy for cell survival [60, 61].
In ischemic brain condition, the research has focused on autophagy in the early stage. It is found that autophagy is the double-edged sword for cerebral ischemia [9, 62]. Excessive autophagy leads to neuronal death which rapidly increases in 3–12 hours after cerebral ischemia [63, 64]. CAPN1, the result of excessive Ca2+ overload, can cleavage beclin-1 lead to defection of autophagosome formation [65]. Moreover, CAPN1 also interrupts the function of Atg protein, the main protein functioning though autophagic process, families resulting in Atg protein dysfunction and autophagic flux impairment [66]. In addition, high level of intracellular Ca2+ and reduced of ATP affects to AMP-activated protein kinase-alpha (AMPKα) phosphorylation, the target of autophagy up-regulation, which inhibits mTOR activation and promote over autophagic flux after ischemic occurring [67]. Incomplete autophagosome formation affects to autophagosome accumulation and trigger neuronal cell death, respectively (Fig. 2).
Lysosomal degradation
Heat shock proteins (Hsp70) are associated with protein folding, complex formation, translocation, and protein degradation. It prevents the abnormal protein formation under stress condition by translocating from the cytosol to the nucleus [68-70]. Under cerebral ischemia, a recent study found that Hsp70 translocates to the luminal side of the lysosome to stabilize the lysosomal membrane [71]. The expression of Hsp70 is significantly increased in 6 hours after ischemic occurring [72-74]. Lysosome, the membrane-bound organelle containing hydrolytic enzyme, is an important component of autophagic process [55]. Under ischemic conditions, lysosome is destroyed by CAPN1 which induces lysosomal membrane permeability resulting to lysosomal breakdown and hydrolytic enzyme releasing into cytoplasm. These can trigger cell death signaling in neuronal cell [75, 76].
In cerebral ischemia, ATP depletion in the brain stimulates anoxic depolarization, resulting in NMDAr overexpression and intracellular Ca2+ accumulation. These changes can activate CAPN1 [22, 77], which translocates to the lysosomal membrane, a change that alters lysosomal membrane permeability, causing the membrane to rupture and cathepsin B release into the cytosol [75, 76, 78]. Moreover, NMDA receptor overexpression provokes nNOS activity, leading to NO generation [79]. Over NO generation is the cause of 4-HNE production resulting from lipid peroxidation [79, 80]. CAPN1 and 4-HNE provoke Hsp70 carbonylation, leading to Hsp70 dysfunction and the loss of lysosomal membrane stability. Consequently, the lysosomal membrane ruptures and cathepsin B is released [71, 81-84]. Cathepsin B release can activate caspase-3 via caspase-11, which leads to apoptosis in the brain (Fig. 2) [85-87].
Therapeutic effects of nmdar antagonist
It is reported that blocking NMDAr by NMDAr antagonist can reduce brain damage from excitotoxicity and intracellular Ca2+ accumulation as neuroprotection in acute stage [38, 88]. The researcher found that ifenprodil, the GluN1 and GluN2B subunits inhibitor, reduced neuronal death in transient cerebral ischemic rat together with contribute phosphorylation of CREB protein as neuroprotective effect [89]. Nimodipine, the VGCC, was used to delay brain damage in early cerebral ischemia. It is found that nimodipine injection via intra-arterial administration protects the brain from cerebral ischemic injury after aneurysmal subarachnoid hemorrhage [90]. Treated with icaritin (ICT), the natural compound extracted from traditional Chinese herb (Epimedium Genus), can protect neuronal cell from glutamate-induced neuronal cell damage by inhibiting DAPK1 and GluN2B activation with promoting phosphorylation of ERK and GluN2A expression, which reduce neuronal cell death in ischemic rats [91].
Regulated autophagy is an important process which can maintain cellular energy and promote cell survival under ischemic conditions [92]. The potential therapeutic treatments for autophagic regulation in cerebral ischemia are interesting. Many researchers reported that regulated autophagy by promoting mTOR activation can reduce brain damage in ischemic condition [93, 94]. Furthermore, the alternative treatment with natural compounds also improves autophagy in ischemia [95]. In glutamate-induced excitotoxic neuroblastoma cell, treated with ST2-104, the nona-arginine (R9)-fused CBD3 peptide, reduce Ca2+ accumulation in cytosol with decrease cell death via Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKKβ) regulation. Moreover, ST2-104 control autophagic process via CaMKKβ/AMPK/mTOR pathway. Likewise, ST2-104 protect the rat brain from transient cerebral ischemic injury after transient cerebral ischemia [96]. Neferine is the alkaloid compound extracted from lotus seeds. It demonstrates the attribute of Ca2+ channel blocker [97], which can moderate intracellular Ca2+ level and regulate autophagic flux via Ca2+-dependent AMPK/mTOR pathway leading to reduce of brain infarction in acute permanent ischemic rats [98].
Blocking NMDAr can reduce neuronal cell damage from glutamate and intracellular Ca2+ toxicity in early cerebral ischemia. However, the severity of ischemic brain varies to occlusion time. Treatment with NMDAr antagonist is suitable for acute cerebral ischemia. The effects of treatment are less efficient to prevent brain damage in long-term occlusion [99, 100].
Conclusion
In the early stage of cerebral ischemia, glutamate excitotoxicity and Ca2+ ions overload play an important role in neuronal dysfunction which can disturb the various cellular physiologies and trigger many downstream cascades of cell death. Autophagy is one of the essential processes which is affected after acute cerebral ischemic occurring. Excessive autophagic flux and lysosomal degradation led to neuronal cell death stimulation. Contrarily, regulated autophagy promotes cell survival and protects the neuron from second damage of ischemia.
 XML Download
XML Download