

 PDF
PDF Citation
Citation Print
Print



The term “multiple primary (MP) cancers” refers to the occurrence of more than one synchronous or metachronous cancer in the same patient [1]. “Synchronous cancers” refer to the occurrence of MP cancers within a six-month timeframe, whereas “metachronous cancers” describe the development of MP cancers with more than a six-month gap between their occurrences [2]. The definition of MP cancers is established based on two guidelines: the criteria of the Surveillance Epidemiology and End Results (SEER) Program [3] and those of the International Association of Cancer Registries (IACR) and the International Agency for Research on Cancer (IARC) [4, 5]. The estimated incidence of MP tumors ranges from 2%–17% [6-9]. Among MP cancer cases, breast and colorectal cancers are the most frequently observed, with the most common tumor combination being breast and colorectal cancer [10]. The combination of MP cancers with hematological malignancies is rare [2].
In patients with MP cancers and hematological malignancies, cases in which second primary cancer is a hematologic malignancy can be confused with therapy-related myeloid neoplasm (T-MN). According to the 4th edition of the WHO diagnostic criteria [11], T-MN is a late complication that occurs following cytotoxic therapies, including chemotherapy and radiotherapy, and includes myelodysplastic neoplasms (MDS), myelodysplastic/myeloproliferative neoplasms (MDS/MPN), and AML, excluding myeloproliferative neoplasm (MPN) or lymphoblastic leukemia. The recent 5th edition of the WHO diagnostic criteria recommends using the term “myeloid neoplasm post cytotoxic therapy” rather than “T-MN” [12].
Studies have demonstrated genetic predisposition in T-MN and MP cancers. Germline variants in genes associated with inherited cancer susceptibility, such as BARD1, BRCA1, BRCA2, CHEK2, TP53, and the Fanconi anemia genes, are identified in approximately 16%–21% of patients diagnosed with T-MN [13-15]. In our previous study involving 53 T-MN patients in Korea, seven patients (13.2%) presented deleterious presumed/potential germline variants in cancer predisposition genes (CPGs), such as BRIP1, CEBPA, DDX41, FANCM, NBN, NF1, and RUNX1 [16]. Using whole-genome sequencing, Whitworth et al. [10] detected pathogenic germline variants in CPGs in 67 (15.2%) out of 440 patients with MP cancers. The commonly mutated CPGs included ATM, BRCA1/2, CHEK2, FH, and PALB2 [10].
We report five patients with MP cancers and hematologic malignancies and describe their clinical characteristics, cytogenetic profiles, and germline and somatic variants. The five patients were identified during our previous study on T-MN at a single institution in Korea [16] and were selected for the present study based on T-MN diagnostic criteria. The patients were diagnosed as having two distinct cancers with more than a six-month gap, and their diagnoses adhered to both the SEER and IACR/IARC MP cancer criteria. Consequently, we classified the patients as having metachronous MP cancers. This study was reviewed and approved by the Institutional Review Board of Seoul National University College of Medicine, Seoul, Korea (IRB No.: 2001-139-1096), and the patients were enrolled with informed consent.
Two patients (MP1 and MP2) had previously been diagnosed with solid cancer without receiving cytotoxic therapy, and their second cancers were MPN (primary myelofibrosis) and MDS, respectively (Table 1). The remaining three patients (MP3, MP4, and MP5) had previously been diagnosed with a hematologic malignancy or solid cancer and had received relevant cytotoxic therapies. In these patients, second hematologic malignancies occurred, which did not manifest as MDS, MDS/MPN, or AML. Cytotoxic therapy reduced the latency for the occurrence of the second tumor: 9–10 years (MP1 and MP2) vs. 1–7 years (MP3, MP4, and MP5). This suggests that genotoxic agents can accelerate the accumulation of mutations and evolution of clones, which are tumorigenic mechanisms [17].
Using bone marrow (BM) aspirates of the five patients obtained after the diagnosis of the second cancer (hematologic malignancies), chromosome analysis, interphase FISH, and targeted next-generation sequencing was performed, as previously described [16]. The only difference from the previous study lay in the variant filtering strategy used: in the present study, only tier I and II variants were selected in somatic mutation analysis, according to the guidelines of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists [18].
Patient MP2 harbored a 17/17p deletion in their second cancer, MDS. Although monosomy 5 or 7, 5q or 7q deletion are commonly observed cytogenetic abnormalities in T-MN, 17/17p deletion is also observed in some T-MN cases [16]. Three patients (MP1, MP3, and MP5) whose second cancers were MPN harbored a JAK2 V617F somatic mutation.
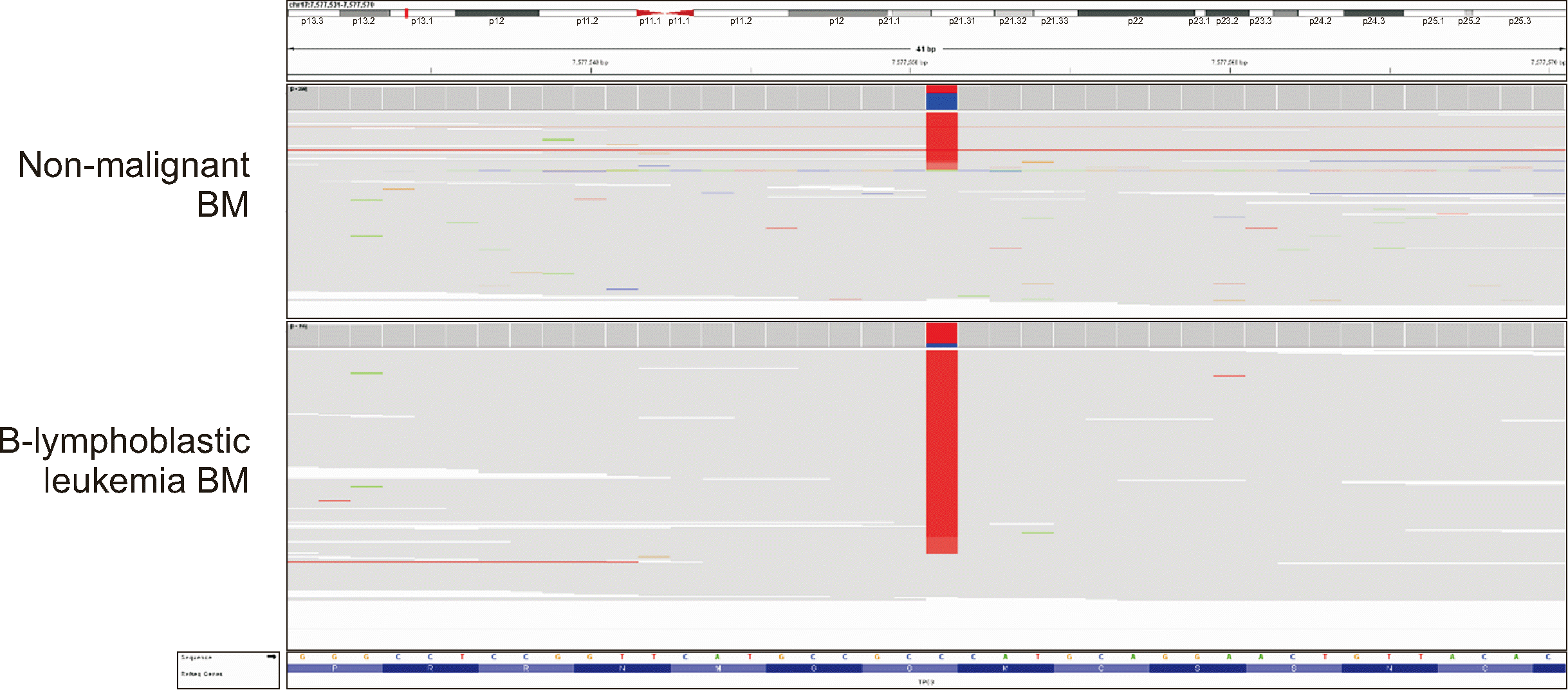
Two patients harbored deleterious potential/presumed germline variants in CPGs (Table 2). Patient MP3 harbored a potential ATM germline variant, which could also be interpreted as a somatic variant owing to the lack of a non-malignant (control) BM sample. The ATM variant was reported in the ClinVar and Human Gene Mutation Database (HGMD) but not in the Catalogue of Somatic Mutations in Cancer (COSMIC), suggesting the possibility of a germline variant. ATM is a BRCA1-associated DNA repair protein [19]. Patient MP4 carried a presumed TP53 germline variant, which was confirmed based on a non-malignant BM sample. Notably, the TP53 variant exhibited different variant allele frequencies (VAFs) of 31.0% and 82.5% in the BM samples from the non-malignant state and B-lymphoblastic leukemia (second tumor), respectively (Fig. 1). This suggests the occurrence of loss of heterozygosity (LOH) in the region covering TP53 c.730. In this case, the tumor suppressor TP53 lost its function owing to LOH, contributing to the tumorigenesis of B-lymphoblastic leukemia. Finally, patient MP4 was diagnosed as having Li–Fraumeni syndrome. Notably, the two patients harboring deleterious potential/presumed germline variants in CPGs were diagnosed with first and second cancers at younger ages than the other three patients, suggesting a potential association between early-age onset and germline predisposition.
In this case series of MP cancers, all second cancers were hematologic malignancies. When managing a patient with a hematologic malignancy and a history of previous cancers, early differentiation of the T-MN is crucial. Otherwise, one should consider the possibility of MP cancers. Despite its limited sample size, our study revealed that two out of five patients (40%) with MP cancers and hematologic malignancies harbored pathogenic germline variants in CPGs. We demonstrated that MP cancers with hematologic malignancies have considerable germline predisposition, as does T-MN. Allogenic hematopoietic stem cell transplantation is the ultimate treatment option for many hematologic malignancies, and related stem cell donors can be considered. Therefore, we strongly recommend that germline variant tests be considered for patients with MP cancers and hematologic malignancies. When germline variants are identified in the patient, screening for detected variants in relatives of patients who are candidates for stem cell donation is advisable.
 XML Download
XML Download