

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Facioscapulohumeral muscular dystrophy (FSHD) is a form of muscular dystrophy that is clinically characterized by asymmetric and progressive muscle weakness involving the face, scapular stabilizers, upper arms, and hip girdle. The disease is predominantly inherited in an autosomal-dominant pattern. However, 10%–30% of cases are attributed to de novo variants, with a high frequency of somatic mosaicism [1-3]. FSHD is caused by aberrant expression of DUX4, which encodes a protein that induces apoptosis by causing immune responses in muscle cells. Each D4Z4 unit on chromosome 4q35 encodes a copy of the DUX4 retrogene [4, 5]. A homologous D4Z4-repeat array has also been identified on chromosome 10, but this region has not been associated with FSHD [6, 7]. Two allelic variations are distal to D4Z4: 4qA and 4qB. Because only the 4qA allele has a polyadenylation sequence, FSHD is only associated with the 4qA allele (the permissive allele) [8].
FSHD is divided into two types according to the cause of aberrant DUX4 expression: FSHD1 and FSHD2 [9]. FSHD1, which accounts for over 95% of FSHD cases, is caused by a contraction of D4Z4 repeats at 4q35 that enables chromatin remodeling and hypomethylation, resulting in DUX4 expression. Healthy people have 11 or more repeats, whereas patients with FSHD1 have fewer than 11 repeats. A minority of patients have FSHD2. These cases show no contraction of D4Z4 repeats but rather have variants in SMCHD1 (18p11.32) or DNMT3B (20q11) that lead to hypomethylation in the D4Z4 region [10, 11].
The number of D4Z4 repeats critically influences the genotype–phenotype correlation [12]. The repeat size correlates positively with the age at onset or diagnosis [13-15] and negatively with disease severity [16]. Penetrance is higher in patients with one to three D4Z4 repeats (85%–100%) than in patients with four to seven D4Z4 repeats (~70%). Patients with few D4Z4 repeats can develop extra-muscular manifestations, such as hearing loss and vascular retinopathy. In contrast, patients with four to seven D4Z4 repeats show moderate symptoms and a later onset than those with fewer repeats. Patients with 7–10 D4Z4 copies do not require wheelchair usage but show high clinical variability.
The standard genetic test method for FSHD1 is Southern blot analysis of EcoRI-digested DNA with the probe, p13E-11. However, this method may yield false-positive results because of nonpathogenic contractions of homologous D4Z4 repeats on 10q26 or the nonpathogenic B-type allele. Although additional restriction enzymes can distinguish between chr4/10 or 4qA/4qB variations [17], this method is labor-intensive and time-consuming. Southern blot analysis is limited to cases where a deletion is proximal to the D4Z4-repeat units [18] or somatic mosaicism is present [19]. Alternative tests, such as optical genome mapping (OGM) [20-22], nanopore sequencing [23], and molecular combing [24], are being researched. OGM is a single-molecule strategy that can be used to construct ordered, genome-wide, high-resolution maps from high-molecular-weight, fluorescently labeled DNA. Applying OGM to diagnose FSHD can overcome the limitations of conventional methods and yield more precise and faster test results. We evaluated the diagnostic performance of OGM in patients with suspected FSHD and their families. Our findings provide the first data regarding the application of OGM for diagnosing FSHD in Korea.
MATERIALS AND METHODS
Patients and Southern blot analysis
We enrolled patients confirmed to have FSHD by Southern blot analysis, family members of patients with FSHD, and patients with clinical signs of FSHD. We defined patients with asymmetric winged scapulae with or without muscle weakness as clinically probable and suspected FSHD cases, respectively. To determine physical disability, we used the 10-grade clinical-severity scale (CSS) developed by Ricci, et al. [25]. The age-corrected CSS (age-corrected CSS=[CSS/age at examination]×1,000) was used to adjust for patient age at examination [26]. This study was approved by the institutional review board (IRB) of Gangnam Severance Hospital, Seoul, Korea (IRB number: 3-2021-0057). Informed consent was waived for this study because participants had already consented to the use of their samples and data for secondary research in another study. Southern blot analysis was performed by external laboratories (Department of Laboratory Medicine, Samsung Medical Center, Seoul, Korea; Department of Neuromuscular Research, National Center of Neurology and Psychiatry, Tokyo, Japan; and Department of Laboratory Medicine, Seoul National University Hospital, Seoul, Korea) using EcoRI- or EcoRI/BlnI-digested genomic DNA and the p13E-11 probe. The number of D4Z4 repeats was determined based on the EcoRI fragment size.
OGM analysis
Ultra-high-molecular-weight (UHMW) genomic DNA (gDNA) was isolated from peripheral blood stored at –80°C following the Bionano Prep SP Frozen Human Blood DNA Isolation Protocol (#30246; Bionano Genomics, San Diego, CA, USA). Cells were pelleted (2,200×g, 2 min) and lysed with proteinase K in a chaotropic buffer after removing the supernatant. After treating the samples with 100 mM phenylmethylsulfonyl fluoride at room temperature (18–25°C) for 10 min (Sigma-Aldrich, Poole, UK), a single Nanobind Disk (Bionano Genomics) and 100% isopropanol were added to the lysate. To increase gDNA binding to the disks, each sample was rotated in a rotator. After three washes using Wash Buffer (Bionano Genomics), each disk was transferred to a new tube, and gDNA was eluted from the disk. The DNA was quantified using a Qubit HS dsDNA Assay Kit and Qubit 3.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA).
Seven hundred fifty nanograms of UHMW gDNA was labeled according to the Bionano Prep Direct Label and Stain (DLS) Protocol (#30206). Sequence-specific labeling was conducted using the Direct Label Enzyme (DLE-1) and DL-Green fluorophore. The labeled DNA was stained blue using DNA-intercalating dye for backbone visualization and loaded on a Bionano Saphyr Chip. Individual DNA molecules were linearized in the nanochannels of the chip and imaged using a Saphyr instrument (Bionano Genomics).
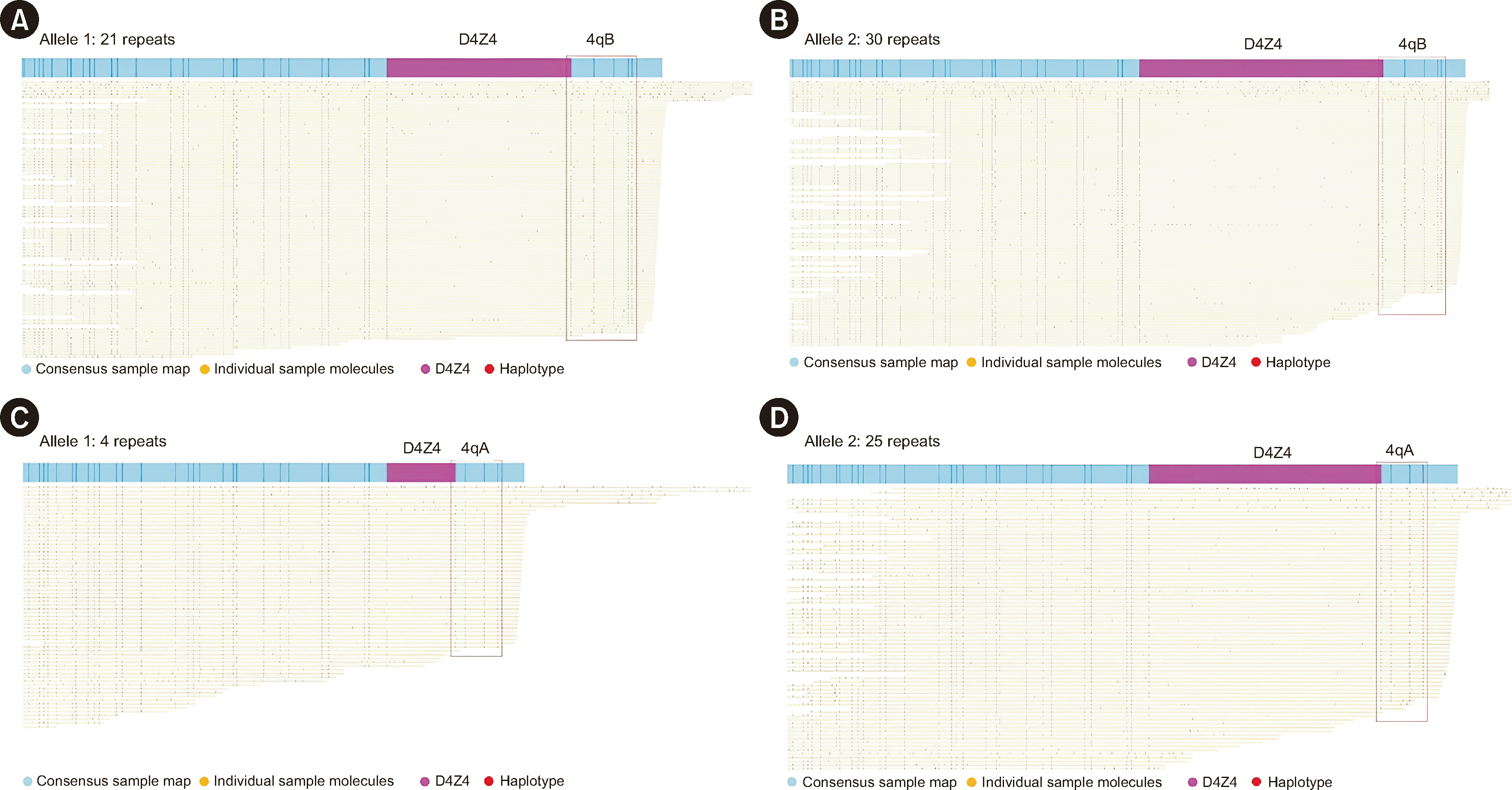
Bionano Solve/Access software was used to size the D4Z4-repeat region and assign haplotypes. The Bionano EnFocus FSHD Analysis pipeline distinguishes haplotypes based on haplotype-specific labeling patterns of molecules aligning with D4Z4-repeat regions for both chromosomes 4 and 10 (hg38). To assess quality and stable regions were assessed (Supplemental Data Table S1). The pipeline calculates the number of D4Z4-repeat units (N) using the following formula: N=(ΔP–Dl–DA|B)/S, where ΔP is the size of the interval between the flanking labels, D1 is the offset for the repeat start, DA and DB are the haplotype-specific offsets for the repeat end, and S is the expected D4Z4-repeat unit size (3.3 Kbp).
Statistical analysis
To analyze whether sex, family history, CSS, age-corrected CSS, and D4Z4-repeat size correlated with the age at symptom onset/diagnosis in our FSHD cohort, we used the Mann–Whitney U-test for categorical variables and Spearman correlation analysis for continuous variables. All P-values were two-tailed, and P<0.05 was considered to reflect a statistically significant difference. Statistical analysis was performed using Analyze-it software v5.68 for Microsoft Excel (Analyze-it Software Ltd., Leeds, UK).
RESULTS
Validation of the OGM method for patients with FSHD1
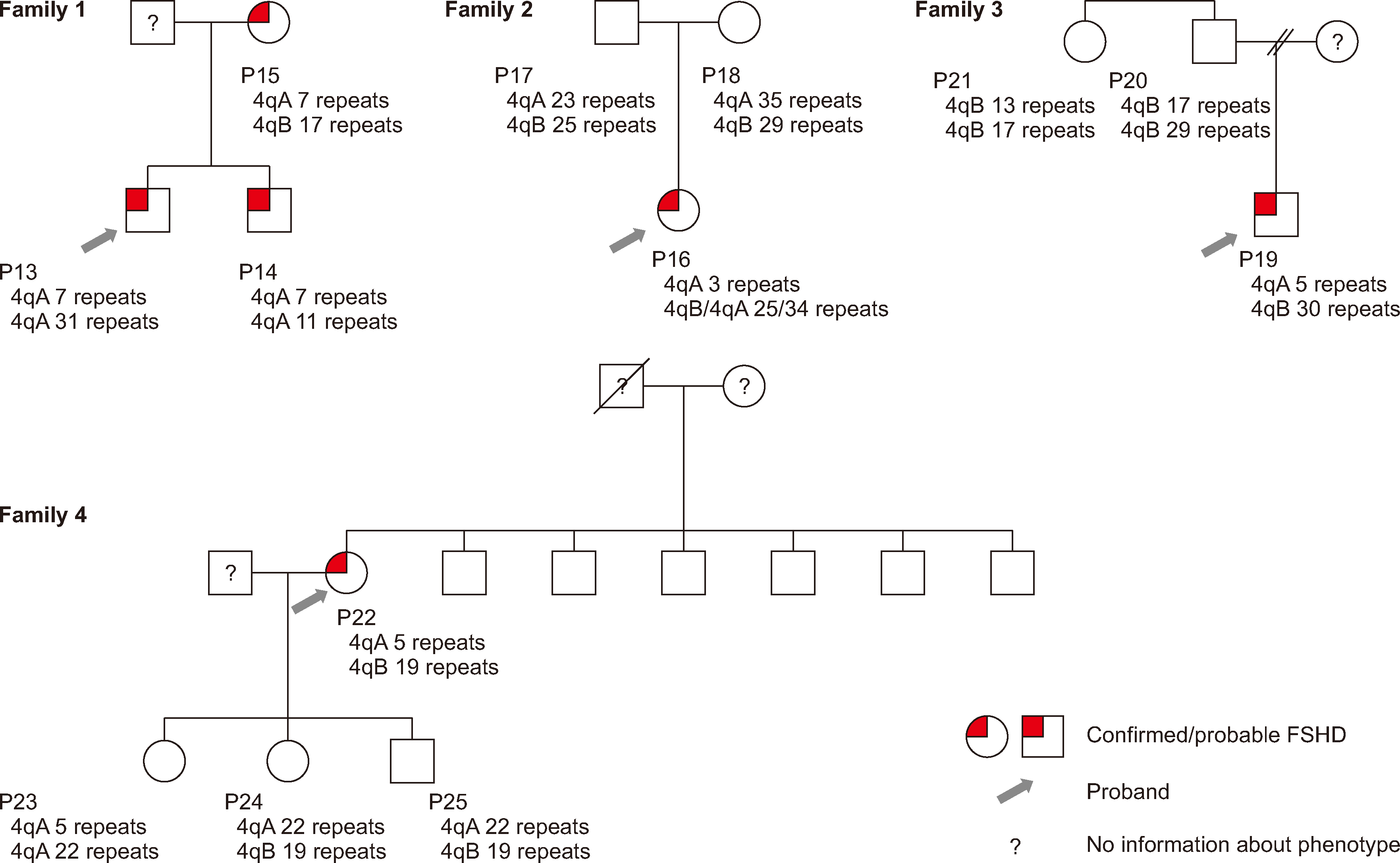
We selected 10 samples (P1, P2, P3, P4, P5, P6, P13, P14, P15, and P22) from patients with a clinically confirmed diagnosis of FSHD1 to analyze the accuracy of OGM. The number of repeats determined by OGM was compared with that determined by Southern blot analysis (Table 1). The OGM results showed 100% concordance with the Southern blot analysis within ±1 repeat. Type A and B haplotype analysis yielded concordant results between Southern blot analysis and OGM with five patients. OGM revealed only one allele in patient P6, suggestive of a homozygous allele; however, we did not perform additional tests, such as a family test. Two of three patients with different repeat sizes found by Southern blotting and OGM (P14 and P15) were family members (Family 1). In terms of the Southern blot results, the allelic configurations in two sons and their mother were inconsistent with Mendelian inheritance; however, OGM confirmed that the repeat size was the same in all family members.
Analysis of FSHD families
We analyzed four families of patients with confirmed/clinically probable FSHD (Fig. 1). In Family 1, to determine whether the same number of D4Z4 repeats could be identified in all family members using OGM, we tested a mother and her two sons with confirmed FSHD. The results confirmed that the pathogenic allele with seven repeats was inherited by both sons from their mother.
Trio analysis was performed for Family 2, with a daughter who showed clinical symptoms. Both parents had nonpathogenic alleles; the cause of FSHD in the daughter appeared to be mosaicism. Two alleles inherited from the parents and one de novo pathogenic allele (three repeats) were identified in the daughter. The repeat-spanning coverage was sufficient to confirm the presence of a mosaic allele (26×). The percentage of reads supporting a pathogenic allele among total reads was 29.9%.
In Family 3, the son showed clinical symptoms of FSHD, and OGM was used to determine the D4Z4-repeat length. The number of repeats was five. A family test was done to determine whether the proband’s condition was familial. No pathogenic allele was identified on the paternal side. Because the parents were divorced, we could not confirm whether the pathogenic allele was maternal or de novo.
In Family 4, genetic testing was performed for the proband’s children (two daughters and one son). A pathogenic allele was identified in daughter 1, who did not present clinical symptoms. Daughter 2 was clinically suspected of having FSHD, but no pathogenic allele was found. To rule out sample switching, we performed a resampling procedure on the two daughters and subsequently repeated the OGM analysis; the results were the same.
Molecular diagnosis of FSHD1 by OGM
We had nine samples available from patients with clinically suspected or probable FSHD and a family member (P7, P8, P9, P10, P11, P12, P16, P19, and P24). To identify the D4Z4-repeat size in each patient, we performed OGM. A decrease in the number of D4Z4 repeats was observed in six patients with clinically probable FSHD (Fig. 2). Among the six patients, two (P7 and P9) were subsequently tested by Southern blotting and showed consistent results, confirming the suspected diagnosis of FSHD.
Correlations among demographics, clinical features, and repeat size
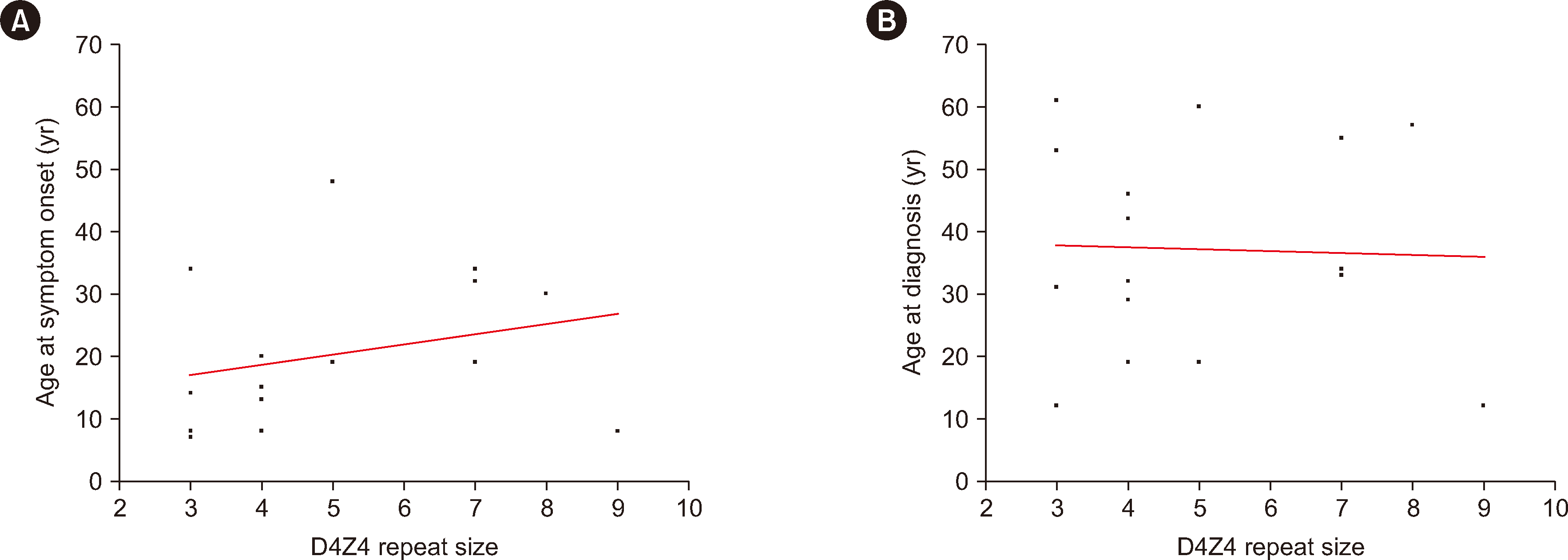
We investigated which factors correlated with the age at symptom onset among our patients. Only one family member was included in each family history analysis. The age-corrected CSS correlated significantly with age at symptom onset (r=–0.545, P=0.029). Sex, family history, CSS, and D4Z4-repeat size were not significantly correlated with age at symptom onset (Fig. 3 and Table 2).
DISCUSSION
We evaluated the clinical utility of OGM for the genetic diagnosis of FSHD1. D4Z4-repeat sizes and haplotypes were successfully determined for all samples tested using OGM. Diagnostic OGM results were concordant with those of Southern blotting in 10 confirmed and two clinically probable cases of FSHD1. OGM is easier and faster to perform than Southern blot analysis (Supplemental Data Table S2). Pathogenic alleles were detected by OGM in six of nine patients with clinically suspected or probable FSHD. OGM can be used effectively for the routine genetic diagnosis of FSHD.
In three cases, OGM revealed one repeat less than Southern blot analysis (one case with four repeats and two cases with seven repeats according to OGM). Previously, the mean size difference between these two methods was 10.1 Kbp, which was attributed to variations in the sizing accuracy of Southern blotting [20]. As Southern blotting is a gel electrophoresis-based method, these discrepancies may be attributed to the compromised sizing quality of the gel. Patients who showed discrepancies in repeat size in this study had less than eight repeats according to both OGM and Southern blot analyses; therefore, there was no issue with the diagnosis of FSHD1. However, caution should be exercised when the repeat count is near the cutoff limit.
We found familial cases with one repeat difference in the OGM results (in Family 2, the proband had 34 repeats, and the mother had 35 repeats; in Family 3, the proband had 30 repeats, and the father had 29 repeats), which has been previously observed [22]. This discrepancy is presumably owing to the conversion of the calculated value of decimal units into the value of the repeat number of integer units.
We identified one de novo case of mosaic FSHD1. Mosaicism likely results from postzygotic D4Z4 contraction in early embryogenesis [19]. Approximately 50% of de novo cases are mosaic, and these patients generally have milder symptoms than patients without mosaicism [19]. In the case of P16, the age at symptom onset was relatively low compared with that of the other patients. However, muscle weakness did not progress during the 2 yr follow-up period. Although we did not perform Southern blot analysis for our patient with mosaicism, mosaicism is often not detected by Southern blot analysis. Our data highlight the diagnostic utility of OGM, as the mosaic form of FSHD1 is challenging to detect. Previously, OGM helped successfully identify cases of mosaicisms at a frequency of 20% based on supporting reads [22] or helped diagnose cases of mosaicism at a higher rate than Southern blotting [20].
Among patients for whom family testing was conducted, the OGM results did not match the phenotype in two cases. In Family 4, daughter 1 had no symptoms but had a pathogenic contraction of the 4qA allele, whereas daughter 2 had symptoms but no pathogenic contraction. A possible cause of the genotype–phenotype discordance in daughter 1 is reduced penetrance. In FSHD, penetrance increases as the D4Z4-repeat number decreases [12]. However, clinical symptoms vary significantly depending on sex and epigenetic, genetic, and environmental factors [12]. In this study, only the age-corrected CSS correlated significantly with age at symptom onset/diagnosis. These results are consistent with previous findings [27]. The correlations between age at symptom onset/diagnosis and repeat size, family history, sex, and CSS were not statistically significant. Previous reports have shown a linear relationship between the D4Z4-repeat size and age at diagnosis/symptom onset [13-15]. Our study did not replicate these findings, which may have been because of the small sample size. Daughter 2 may have FSHD2 due to hypomethylation of the D4Z4-repeat array from other causes. With Bionano EnFocus FSHD analysis, both the D4Z4 region and copy-number variants of SMCHD1 can be analyzed. However, sequence variants of SMCHD1 and DNMT3B, which are associated with other causes of FSHD2, were not assessed in previous studies [10, 11].
This study had several limitations. First, it was a single-center study with a limited sample size. Furthermore, OGM cannot be used as a clinical diagnostic test in Korea, despite a study reporting diagnostic performance results after routine implementation of OGM as a frontline diagnostic test method [20]. In Korea, Southern blotting is the only approved method for clinical FSHD testing; however, very few clinical laboratories conduct Southern blot analysis, leading to challenges in patient diagnosis and management. In this study, some suspected or probable FSHD cases without pathogenic contraction found by OGM were not confirmed using Southern blot analysis. However, the OGM results were concordant with the Southern blot results in all cases. Additional patient and pedigree analyses explained the clinical symptoms and family history, indicating that the OGM results were trustworthy. Finally, we compared the results and time required for OGM and Southern blotting, but we did not analyze the material costs (equipment and reagents). OGM has comparable accuracy to Southern blotting and requires less time and effort. Nevertheless, the cost of purchasing equipment and reagents may be higher than that for Southern blotting, which should be considered for routine implementation.
We evaluated the clinical applicability of OGM as a diagnostic method for FSHD. Our results indicate that OGM enabled accurate and reliable detection of D4Z4-repeat array abnormalities. OGM requires substantially less time and effort than conventional Southern blotting. OGM is a valuable tool for the genetic diagnosis of FSHD.
 XML Download
XML Download