

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Tumor immune escape is a common feature and hallmark of primary malignant tumors. In the tumor microenvironment (TME), cancer cells can evade immune surveillance by infiltrated cytotoxic lymphocytes, including CD8+ T and natural killer (NK) cells, which is induced via interaction between the immune checkpoint molecules programmed cell death 1 (PD-1, encoded by PDCD1) on cytotoxic lymphocytes and programmed cell death ligand 1 (PD-L1, encoded by CD274) on cancer cells [1, 2]. PD-L1/PD-1 signaling inhibits the antitumor activities of tumor-infiltrating cytotoxic lymphocytes, resulting in enhanced immune evasion-induced tumor growth [3]. PD-L1 is upregulated in many human tumor types [3, 4] and is correlated with poor prognosis [5, 6]. PD-L1 expression in the TME can be upregulated at the transcriptional, post-transcriptional, and post-translational levels by numerous factors, including inflammatory stimuli and oncogenic pathways [3]. We have previously reported that β-catenin transactivation, which has been detected in various cancer types [7], can induce the transcriptional upregulation of CD274 in an AKT-mediated β-catenin S552 phosphorylation-dependent manner [4]. In addition, PD-1 levels are elevated in tumor-infiltrating cytotoxic lymphocytes in many TME types compared with those in normal tissues [8-10]. However, the precise mechanisms by which the TME upregulates PD-1 expression remain unknown.
Metformin is the most widely used drug for treating type 2 diabetes. Epidemiological studies have shown that patients with diabetes treated with metformin have reduced cancer and cancer-related mortality risks [11]. Accumulating evidence from preclinical studies shows that metformin exerts antitumor effects [12]. Moreover, metformin reportedly enhances the sensitivity to radiotherapy [13] and chemotherapy [14], particularly when standard chemotherapeutic agents are used at suboptimal doses [15]. Metformin inhibits mitochondrial complex I of the respiratory chain and causes energy depletion, which activates adenosine monophosphate-activated protein kinase (AMPK) [16], a major sensor of the energy status of cells, and subsequently inhibits the mechanistic target of rapamycin [17], thus restraining cancer growth. Metformin also elicits anticancer effects independent of AMPK [18, 19]. Metformin reportedly promotes antitumor immunity by downregulating PD-L1 expression in several cancer cell types [20-22]. However, most studies on the antitumor immune activity of metformin focused on PD-L1 expression in cancer cells, not on PD-1 expression in infiltrating cytotoxic lymphocytes within the TME. In addition, the detailed molecular mechanisms of metformin-mediated antitumor immunity are not well understood.
We aimed to investigate the mechanisms by which the TME upregulates PD-1 expression in tumor-infiltrating cytotoxic lymphocytes and whether metformin can reprogram the tumor immune microenvironment by modulating immune checkpoint expression (i.e., PD-L1 and PD-1), and to elucidate the underlying mechanisms. We hypothesized that metformin can downregulate both PD-L1 expression in cancer cells and cancer-induced PD-1 expression in tumor-infiltrating cytotoxic lymphocytes and thus may induce antitumor immunity.
MATERIALS AND METHODS
Luciferase reporter gene assay
Transcriptional activities of TCF/LEF-1 in A431 cells were measured as described previously [23]. Relative luciferase activity levels were normalized to those in untreated cells or that in the Renilla control plasmid. Each assay was performed at least three times.
NK cell-mediated tumor cell cytotoxicity assay
A431 cells were used as target cells, and human primary NK cells (purified from cord blood of healthy donors in Dong-A University Hospital, Busan, Korea [2-1040709-AB-N-01-202303- BR-002-02]) were used as effector cells. Before inducing cytotoxicity, A431 cells and NK cells were pretreated or not with metformin for 24 hrs. The A431 cells were then labeled with carboxyfluorescein succinimidyl ester (CFSE; CFSE Cell Division Tracker Kit, #423801; BioLegend, San Diego, CA, USA) at 37°C for 30 mins. Target cell cytotoxicity was initiated by coculture with NK cells at an effector:target (E:T) ratio of 1:1 for 4 hrs. After coculture, the A431 cells were harvested and stained with Fixable Viability Dye (FVD; #65-0865-14; Invitrogen, Waltham, MA, USA). After 20 mins, the cells were washed and subjected to flow-cytometric analysis. CFSE/FVD-double-positive cells were regarded as dead cells.
Animal studies
C56BL/6 mice were obtained from OrientBio (Seongnam, Korea) and were housed in the animal facility of Dong-A University. All experiments were performed in accordance with relevant institutional and national guidelines. All animal procedures and maintenance conditions were approved by the Dong-A University Institutional Animal Care and Use Committee (DIACUC-21-48). We subcutaneously injected 5×105 B16F10 murine melanoma cells into five-week-old male C56BL/6 mice (N=5 mice/group). On day 3 post-tumor implantation, 200 mg/kg of metformin dissolved in phosphate-buffered saline (PBS) was intraperitoneally injected daily. Mice in the control group were injected with the same volume of PBS. At 20 days after tumor implantation, the mice were sacrificed by carbon dioxide inhalation to analyze tumor volume and weight, followed by flow-cytometric analysis.
Other methods
Materials, cell culture, transfection, DNA constructs, quantitative real-time PCR analysis, immunoprecipitation and immunoblotting analysis, ubiquitylation assay, preparation of conditioned medium (CM), and flow cytometric analysis are available in the supplemental materials.
Statistical analysis
All quantitative data are presented as the mean±SD of at least three independent experiments. The normality of the data was assessed using Shapiro-Wilk test. Means of two groups were compared using Student’s t-test. Means of more than two groups were compared using one-way ANOVA, followed by Tukey post-hoc tests. Normality and homogeneity assumptions were met. The SPSS statistical package version 12 (SPSS Inc., Chicago, IL, USA) was used for all statistical analyses. Values of P<0.05 indicated statistically significant differences.
Results
Metformin inhibits both PD-L1 expression in cancer cells and PD-1 expression in infiltrating immune cells in the TME
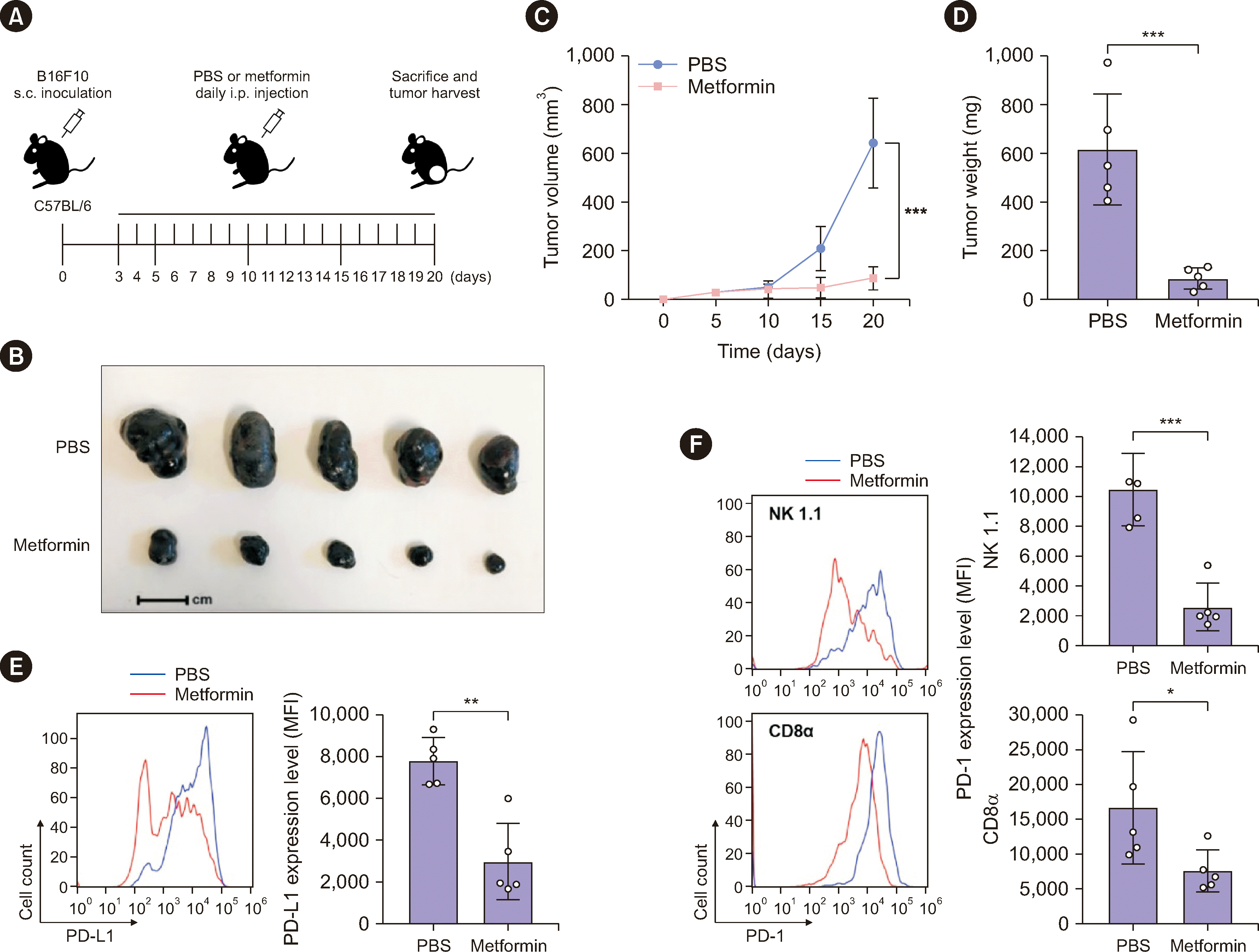
To determine the effects of metformin on immune checkpoints in the TME, C57BL/6 mice were subcutaneously implanted with B16F10 murine melanoma cells, followed by intraperitoneal administration of metformin (Fig. 1A). We did not observe any abnormalities in mouse body weight or behavior. However, metformin effectively exhibited anticancer activity in B16F10 cell-harboring mice, as reflected by decreases in tumor volume and weight (Fig. 1B–1D). B16F10 tumors were extracted, dissociated to obtain single-cell suspensions, and subjected to flow-cytometric analysis. As shown in Fig. 1E, metformin significantly suppressed PD-L1 expression on the surface of B16F10 cancer cells. Consistent with previous findings [8-10], PD-1 expression on the surfaces of tumor-infiltrating NK and CD8+ T cells was significantly upregulated compared with that on splenic naïve NK and CD8+ T cells (Supplemental Data Fig. S1A). Notably, we found that metformin also suppressed PD-1 expression on the surfaces of B16F10 tumor-infiltrating NK and CD8+ T cells (Fig. 1F).
Based on the inhibitory effects of metformin on both PD-L1 and PD-1 expression, we performed an NK cell killing assay using human primary NK cells and A431 human epidermoid carcinoma cells. We cocultured metformin-pretreated A431 cells with metformin-pretreated human primary NK cells and found that metformin treatment significantly induced NK cell-mediated cancer cell death compared with that in the untreated control (Supplemental Data Fig. S1B). These results suggest that metformin downregulates PD-L1 expression on cancer cells and PD-1 expression on infiltrating immune cells (NK and CD8+ T cells) to reduce tumor cell immune evasion in the TME.
Metformin inhibits the transcriptional expression of PD-L1 in an AMPK-downregulated AKT/β-catenin signaling-dependent manner
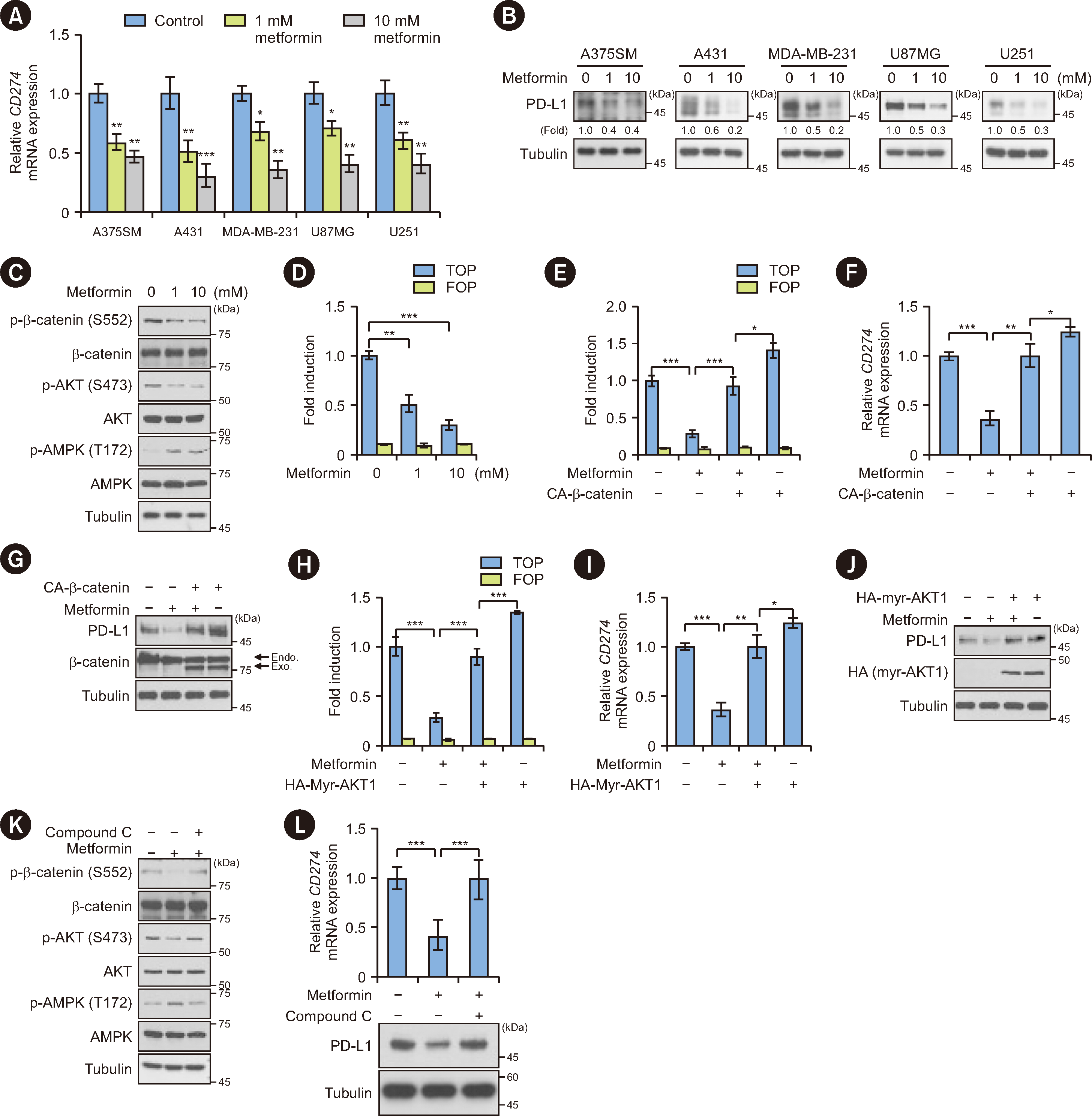
To determine the mechanism by which metformin downregulates PD-L1 expression on cancer cells, we treated A375SM human melanoma cells, A431 human epidermoid carcinoma cells, U87MG and U251 glioblastoma (GBM) cells, and MDA-MB-231 human breast carcinoma cells with metformin. Quantitative PCR and immunoblotting analyses showed that metformin treatment significantly inhibited CD274 (encoding PD-L1) mRNA expression (Fig. 2A) and PD-L1 protein expression (Fig. 2B) in a dose-dependent manner.
We previously reported that AKT-mediated β-catenin S552 phosphorylation is instrumental for β-catenin transcriptional activity to enhance CD274 transcription [4]. Therefore, we investigated the effect of metformin on AKT/β-catenin signaling. As shown in Fig. 2C, metformin increased AMPK Thr172 phosphorylation, which is consistent with previous findings [16]. It significantly inhibited AKT S473 phosphorylation and β-catenin S552 phosphorylation in a dose-dependent manner. In line with these results, metformin treatment suppressed β-catenin transcriptional activity (TOP-FLASH) in A431 cells (Fig. 2D). Notably, the inhibitory effects of metformin on β-catenin transcriptional activity (Fig. 2E and 2H), CD274 mRNA expression (Fig. 2F and 2I), and PD-L1 protein expression (Fig. 2G and 2J) were abrogated upon expression of a constitutively active (CA-) β-catenin mutant or myristoylated AKT1 (myr-AKT1) in A431 cells. These results demonstrate that metformin inhibits the transcriptional expression of PD-L1 by downregulating AKT/β-catenin signaling.
AMPK is a negative regulator of the AKT pathway in cancer cells [24]. Ectopic expression of CA-AMPK inhibited AKT S473 phosphorylation, resulting in decreased β-catenin S552 phosphorylation and PD-L1 expression in A431 cells (Supplemental Data Fig. S2). We next examined whether the suppressive effect of metformin on AKT/β-catenin signaling was mediated via AMPK activation. Metformin-inhibited AKT S473 phosphorylation and β-catenin S552 phosphorylation (Fig. 2K) and subsequent PD-L1 expression (Fig. 2L) were significantly reversed upon treatment with compound C, a pharmacological inhibitor of AMPK. These results suggest that metformin inhibits PD-L1 expression in an AMPK-reduced AKT/β-catenin signaling-dependent manner.
Cancer cells enhance PD-1 protein stability in NK and T cells
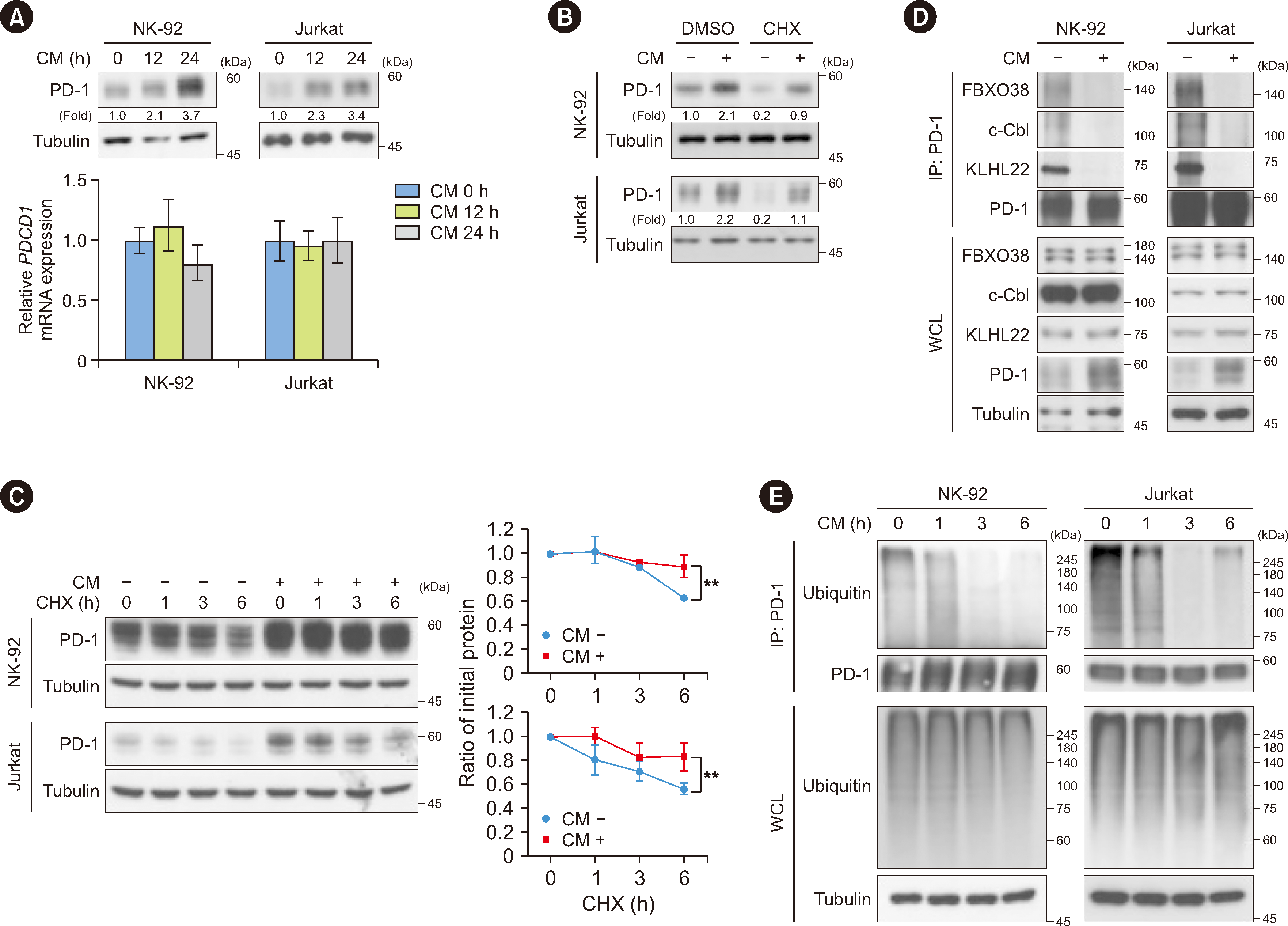
Consistent with previous reports showing that PD-1 levels are markedly elevated in tumor-infiltrating lymphocytes in many TME types compared with those in normal tissues [8-10], we observed an increase in PD-1 expression in tumor-infiltrating NK and CD8+ T cells within B16F10 tumors (Fig. 1). However, the specific molecular mechanisms remain largely unknown. Tumor-derived soluble factors within the TME provide an immunosuppressive environment [25]. To identify the role of cancer-secreted factors in PD-1 expression on immune cells, we collected CM from A431 or B16F10 cells and then treated NK-92 natural killer cells, Jurkat cells, mouse primary splenocytes, and human primary NK cells with the CM. Quantitative PCR and immunoblotting analyses showed that tumor CM treatment substantially enhanced PD-1 protein expression (Fig. 3A upper panel and Supplemental Data Fig. S3 upper panel), whereas no significant alterations were detected in the mRNA expression of Pdcd1 (encoding PD-1) in these four cell types (Fig. 3A lower panel and Supplemental Data Fig. 3 lower panel). To determine whether the tumor CM-induced increase in PD-1 expression resulted from increased PD-1 stability, we pretreated NK-92 and Jurkat cells with cycloheximide (CHX) to block protein synthesis. CHX did not reduce the fold induction of PD-1 expression upon CM treatment (Fig. 3B). In addition, tumor CM treatment increased the half-life of endogenous PD-1 (Fig. 3C). These results suggest that tumor CM enhances PD-1 expression primarily by enhancing PD-1 stability.
Several E3 ubiquitin ligases, including FBXO38 [26], c-Cbl [27], and KLHL22 [28], target PD-1 protein for degradation. To determine the effect of tumor CM on the association between corresponding E3 ligases and PD-1, we performed co-immunoprecipitation assays and found that tumor CM treatment significantly disrupted the binding of endogenous PD-1 to all three endogenous E3 ligases without affecting the expression levels of the E3 ligases (Fig. 3D). Concordantly, ubiquitylation assays revealed that tumor CM treatment decreased PD-1 polyubiquitylation in NK-92 and Jurkat cells (Fig. 3E). These results suggest that tumor-derived soluble factors induce PD-1 stability via dissociation of PD-1 from E3 ligases.
Metformin inhibits cancer-enhanced PD-1 expression via AMPK-dependent deregulation of PD-1 protein stability
We next determined the effect of metformin on tumor CM-induced PD-1 expression. We found that tumor CM-induced increases in PD-1 protein levels were significantly suppressed by metformin in NK-92 and Jurkat cells, mouse primary splenocytes, and human primary NK cells (Fig. 4A and Supplemental Data Fig. S4). To determine the mechanism underlying the metformin-induced PD-1 protein expression suppression, we treated NK-92 and Jurkat cells with a proteasome inhibitor, MG132, which blocked metformin-reduced PD-1 protein expression (Fig. 4B), indicating that metformin induced proteasomal degradation of PD-1 protein. Concordantly, metformin treatment reduced the CM-induced increase in the half-life of endogenous PD-1 in NK-92 and Jurkat cells (Fig. 4C). Furthermore, co-immunoprecipitation and ubiquitylation assays showed that metformin treatment reversed the tumor CM-induced reduction in the binding of endogenous E3 ligases (FBXO38, c-Cbl, and KLHL22) to endogenous PD-1 (Fig. 4D) and PD-1 polyubiquitylation (Fig. 4E), whereas it did not affect the expression levels of the E3 ligases (Fig. 4D lower panel).
To address the specific role of AMPK in PD-1 expression, 293T cells were cotransfected with PD-1 and CA-AMPK. The results revealed that AMPK activation abolished the tumor CM-induced increase in PD-1 expression and decrease in PD-1 polyubiquitylation (Fig. 4F). Moreover, the level of PD-1 polyubiquitylation was decreased in AMPKα1/2 double-knockout (DKO) mouse embryonic fibroblasts (MEFs) compared with that in wild-type (WT) AMPK-expressing MEFs (Fig. 4G). These findings suggested that AMPK negatively regulates PD-1 protein stability. Therefore, we next determined whether the inhibitory effect of metformin on PD-1 expression would be mediated by AMPK activation. Treatment with a pharmacological inhibitor of AMPK compound C blocked the metformin-induced decrease in PD-1 expression (Fig. 4H) and increase in PD-1 polyubiquitylation (Fig. 4I) in NK-92 and Jurkat cells. Collectively, these results indicate that metformin inhibits tumor-derived soluble factor-induced PD-1 expression via AMPK-dependent downregulation of PD-1 protein stability.
DISCUSSION
The representative immune checkpoint molecules PD-L1 and PD-1 are overexpressed in the TME. They play a crucial role in tumor immune evasion, resulting in enhanced tumor growth and a poor prognosis [3]. Therefore, the blockade of PD-L1/PD-1 signaling is recognized as an attractive strategy for cancer immunotherapy. Therapeutics targeting PD-L1/PD-1 signaling result in tumor regression in several cancer types, including melanoma and lung cancer [2, 3]. However, the therapeutic effectiveness of PD-L1/PD-1 antagonists against solid tumors is currently unsatisfactory. Only a portion of patients respond to immunotherapy [3]. This hurdle must be overcome, and alternative agents must be explored to expand their benefits and improve their efficacy and, consequently, patient prognosis. Recent studies have shown that metformin treatment alone can enhance antitumor immunity [20-22]. In addition, metformin synergizes with immune checkpoint inhibitors, as shown in clinical and preclinical studies [20, 21, 29]. However, the exact molecular mechanism of metformin-mediated antitumor immunity remains unclear.
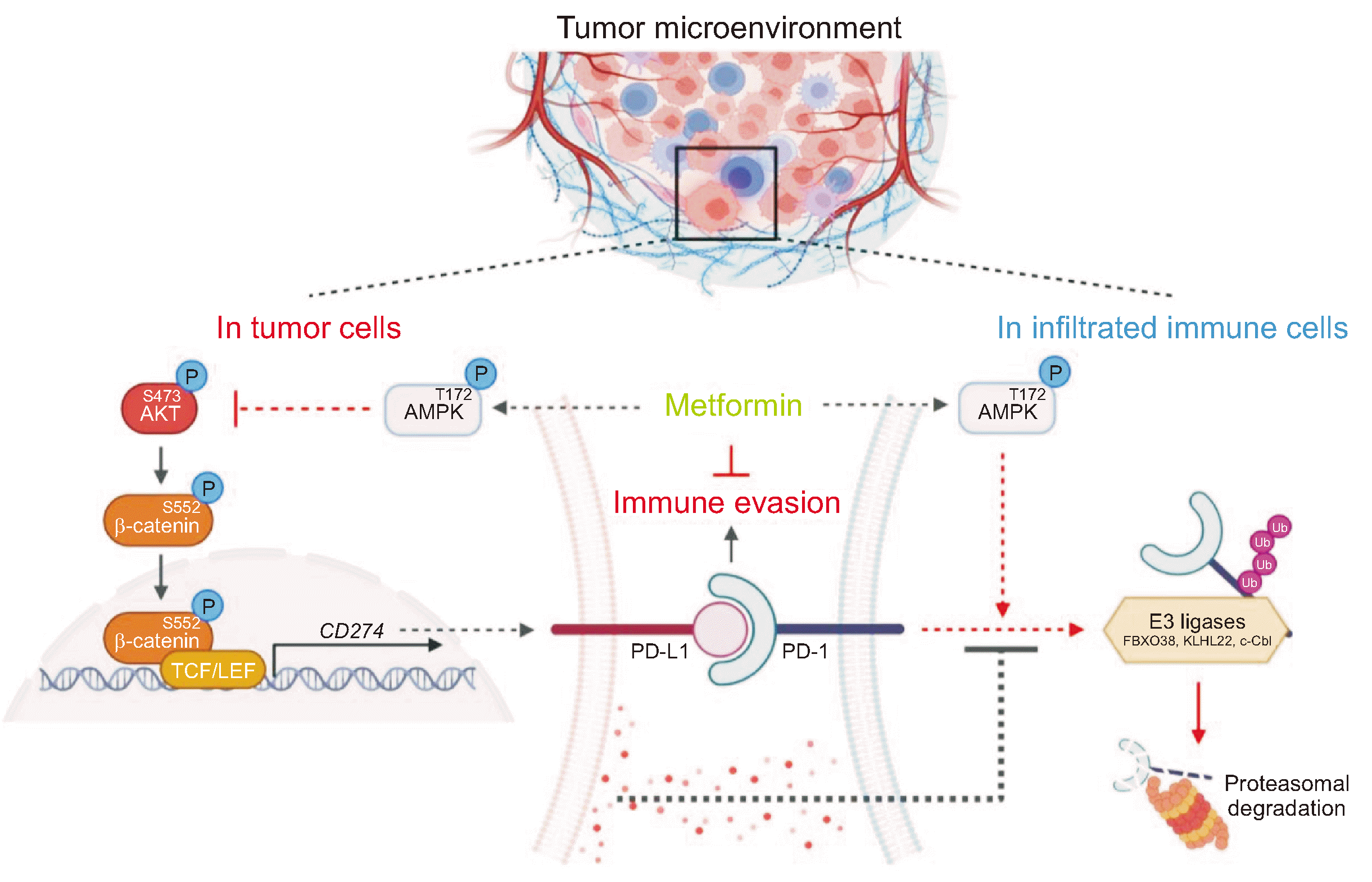
We demonstrated that metformin downregulated the transcriptional expression of PD-L1 via inhibition of the AKT/β-catenin axis in cancer cells and inhibited tumor-derived soluble factor-mediated promotion of PD-1 stability by enhancing the association of PD-1 with E3 ligases and PD-1 polyubiquitylation in tumor-infiltrating cytotoxic lymphocytes. These inhibitory effects of metformin on both PD-L1 and PD-1 expression are AMPK activation-dependent (Fig. 5). Our findings provide a deeper understanding of the antitumor-responsive roles of metformin within the TME, along with new regulatory mechanisms.
Consistent with previous reports showing high PD-1 expression on tumor-infiltrating cytotoxic lymphocytes in many human TME types [8-10], our study showed robust PD-1 expression on the surfaces of tumor-infiltrating NK and CD8+ T cells in the B16F10 tumor model. Numerous studies have shown that various signaling pathways and transcription factors are involved in the induction of PD-1 transcription in several immune cell types [30-33]. T-cell receptor (TCR) activation can induce PDCD1 transcription via NFATc1 activation in CD8+ T lymphocytes [30]. Interleukin (IL)-12 and IL-6 signals promote PD-1 transcription by activating the STAT3/STAT4 pathway upon TCR activation [31]. Interferon-α per se or synergistically with TCR can upregulate PD-1 expression by activating JAK/STAT signaling [32, 33]. Recent studies have shown that PD-1 can be regulated at the post-translational level; PD-1 undergoes ubiquitination by E3 ubiquitin ligases, including FBXO38 [26], c-Cbl [27] and KLHL22 [28], followed by proteasomal degradation. The present study revealed that PD-1 expression is upregulated at the post-translational level, not the transcriptional level, upon interaction with a tumor. Many types of cells and soluble factors within the TME can induce the dysfunction of antitumor immune-responsive cells to provide an immunosuppressive environment, resulting in tumor immune escape. Small molecules such as cytokines (e.g., IL-6 and IL-10), growth factors (e.g., vascular endothelial growth factor and transforming growth factor-beta), and metabolites (e.g., lactic acid) secreted by cancer cells are involved in the process [25, 34]. We found that tumor-derived soluble factors induce the dissociation of PD-1 from all types of ubiquitin E3 ligases, resulting in reduced PD-1 polyubiquitylation and enhanced PD-1 protein stability. However, the comprehensive mechanisms underlying tumor-derived soluble factor-induced dissociation of PD-1 from E3 ligases and types of tumor-derived soluble factors responsible for PD-1 stability mediated by the mechanism remain to be explored.
The PD-L1/PD-1 pathway, as an important immune checkpoint signaling pathway, can inhibit the activation of tumor-infiltrating cytotoxic lymphocytes and enhance the immune tolerance of cancer cells, thereby achieving tumor immune evasion [3]. Metformin can reprogram the tumor immune microenvironment to shift from a protumoral state toward a more antitumoral state. We and others have shown that metformin can inhibit PD-L1 expression at the transcriptional and post-translational levels in many cancer cell types [20-22]. Metformin plays a key role in maintaining the high multifunctional capacity of tumor-infiltrating lymphocytes, including the infiltration and activation of CD8+ cytotoxic T lymphocytes in tumor tissues [35, 36]. Moreover, metformin can enhance both the number and activity of peripheral NK cells, as well as tumor infiltration, thereby benefiting patients with cancer from NK cell-mediated cytotoxicity [37]. The present study revealed that metformin suppresses tumor-derived soluble factor-induced PD-1 expression in tumor-infiltrating cytotoxic lymphocytes. Collectively, previous and our current findings imply that metformin may exert an antitumor immune effect in part by downregulating both PD-L1 and PD-1 expression in the TME.
In summary, our findings revealed that metformin decreases AKT-mediated β-catenin S552 phosphorylation and subsequent β-catenin transactivation to inhibit the transcriptional expression of PD-L1 in human cancer cells. We demonstrated for the first time that metformin decreases PD-1 protein stability in tumor-infiltrating cytotoxic lymphocytes by blocking the tumor-induced reduction in the binding of PD-1 to E3 ligases and PD-1 polyubiquitylation. Our findings suggest that metformin blocks both PD-L1 and PD-1 within the TME to exert an antitumor-immune effect, expanding our understanding of the detailed antitumor-responsive roles of metformin and revealing new regulatory mechanisms. Therefore, metformin can be used in combination therapy to improve the response rate in immunotherapy.
 XML Download
XML Download