

 PDF
PDF Citation
Citation Print
Print



Highlights
· EAT, easily assessed via imaging, is anatomically and functionally connected to the myocardium.
· Cytokines and fatty acids from EAT worsen myocardial remodeling, causing HF.
· In HFrEF, reduced EAT volume indicates metabolic dysfunction.
· Conversely, in HFpEF, higher EAT levels correlate with adverse hemodynamic profiles.
· EAT may serve as a target for HF therapies.
INTRODUCTION
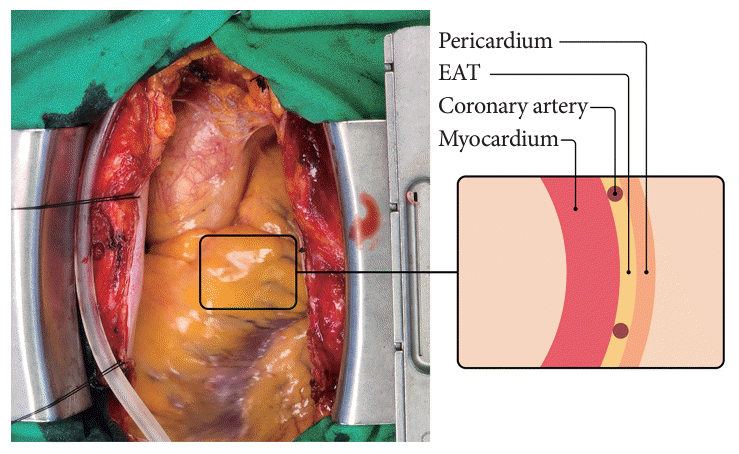
The epicardial adipose tissue (EAT) is a visceral adipose tissue (VAT) located in the heart [1]. With an increased interest in organ-specific adiposity, a close association between EAT and heart failure (HF) has been suggested because of its anatomical proximity to the heart. EAT is located in the interventricular or atrioventricular groove and surrounds nearly all coronary arteries, constituting 15% to 20% of the heart mass [2]. Fig. 1 shows the gross anatomy of EAT in an 81-year-old woman with threevessel coronary artery disease (CAD) who underwent a coronary artery bypass graft. EAT has unique characteristics compared to those of the pericardial adipose tissue (PAT), as EAT is located between the myocardium and visceral pericardium, has an embryonic origin from the splanchnopleuric mesoderm, and is supplied by branches of the coronary artery. In contrast, PAT originates from the primitive thoracic mesenchyme and is vascularized by branches of the internal mammary artery [3]. EAT and the myocardium share the same blood supply, and there is no fascia separating them histologically, making them anatomically and functionally contiguous [4]. Some studies do not differentiate between EAT and PAT, describing PAT as a broader concept that includes EAT. However, this study specifically concentrates on EAT as a true VAT of heart [5].
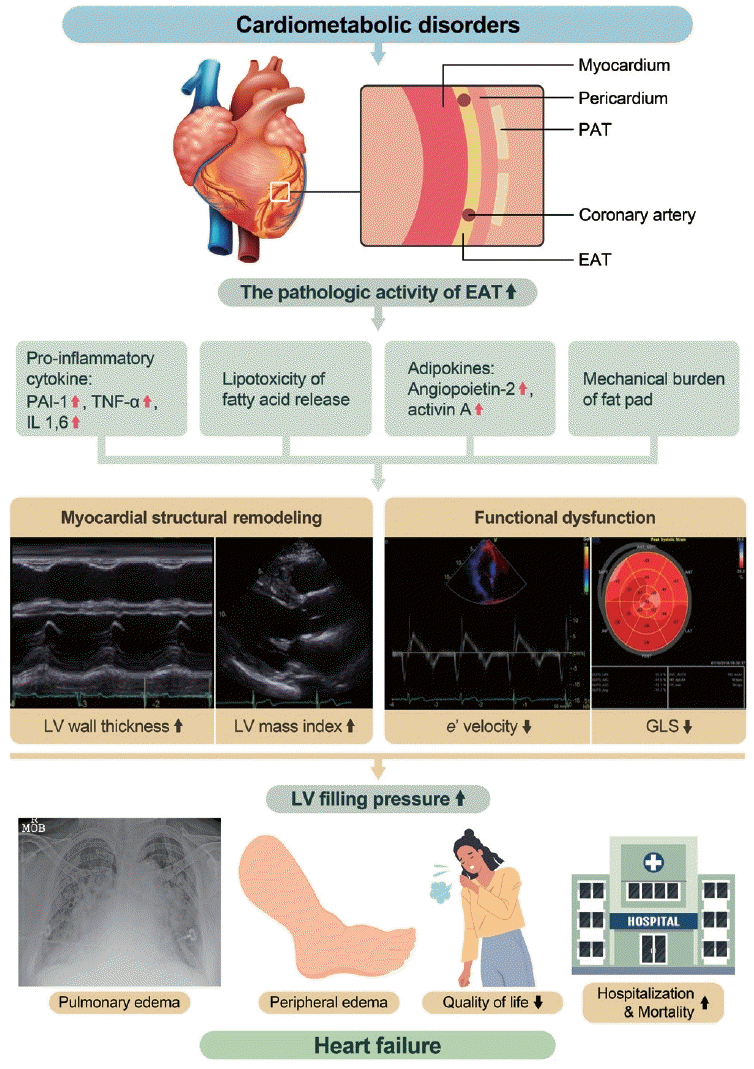
EAT is a bioactive organ that secretes several adipokines, including pro-inflammatory and pro-fibrotic cytokines, which can induce myocardial remodeling and dysfunction [6]. In pathological conditions, such as obesity, EAT releases excessive free fatty acids (FFAs) from its lipid store, which can induce lipotoxic effects on the myocardium [7]. The close proximity of EAT to the myocardium and its secretion of pathological cytokines and FFAs have led to investigations into the role of EAT in the development and progression of HF [8].
Despite the dramatic improvement in short- and long-term outcomes of HF with established guideline-directed medical therapy, HF remains a significant health burden worldwide with increasing prevalence [9-13]. Current HF guidelines emphasize the importance of individualized assessment and management according to HF phenotypes [14-16]. Obesity is a wellknown risk factor for HF, as it is associated with a high-volume load, impaired insulin resistance, and metabolic disturbances [17]. Several surrogate markers of adiposity reflect the risk of HF. In this review, we discuss the pathological role of EAT on the myocardium and its association with HF, as well as the possibility of EAT as a therapeutic target in HF.
ASSESSMENT OF EAT
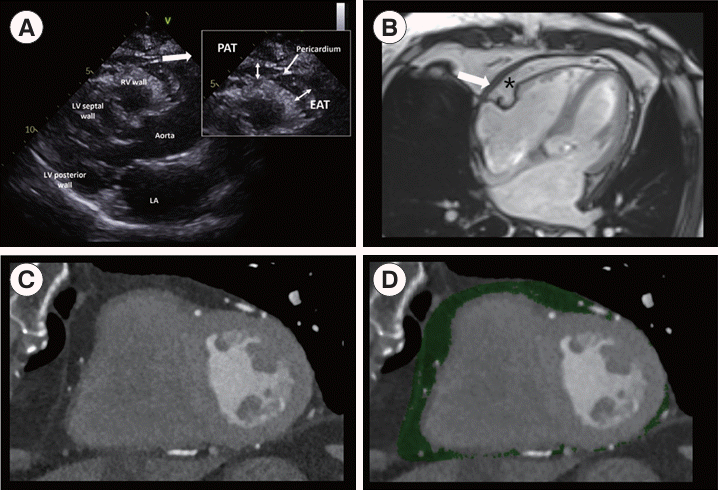
In the current era of multi-modal imaging, various tools are available to assess the amount and location of EAT. Echocardiography is a non-invasive and relatively inexpensive technique that can easily evaluate the amount of EAT [4,18]. The presence of EAT between the myocardium and the visceral layer of the pericardium can be identified by echocardiography. Representative echocardiographic images of EAT are shown in Fig. 2. Echocardiographic EAT thickness is typically defined as the maximal thickness during end-systole [2,19,20] and is measured perpendicular to the right ventricle (RV) free wall of the aortic annulus in the parasternal long-axis view. Echocardiographic EAT thickness is a well-validated and reproducible marker in the general population and in patients with various cardiovascular diseases (CVDs), with EAT thicknesses ranging from 1 to 23 mm [1,21,22]. In a study of a large population, mean EAT thickness values were found to be 7 mm in men and 6.5 mm in women [4]. However, echocardiography only provides a linear measurement of EAT; thus, the amount of EAT evaluated by this method may be less accurate than that measured by computed tomography (CT) or magnetic resonance imaging (MRI) [8]. Although CT and MRI may be more expensive and invasive techniques, they provide a more accurate evaluation of the amount of EAT by volumetric or area assessment [23,24]. Additionally, using CT and MRI, characterization of EAT at any location with higher resolution can be achieved, thus providing more location-specific implications of EAT. For example, EAT around the left atrium (LA) is closely associated with atrial fibrillation and the LA volume index [25]. The characteristics of each imaging modality for evaluating EAT are summarized in Table 1.
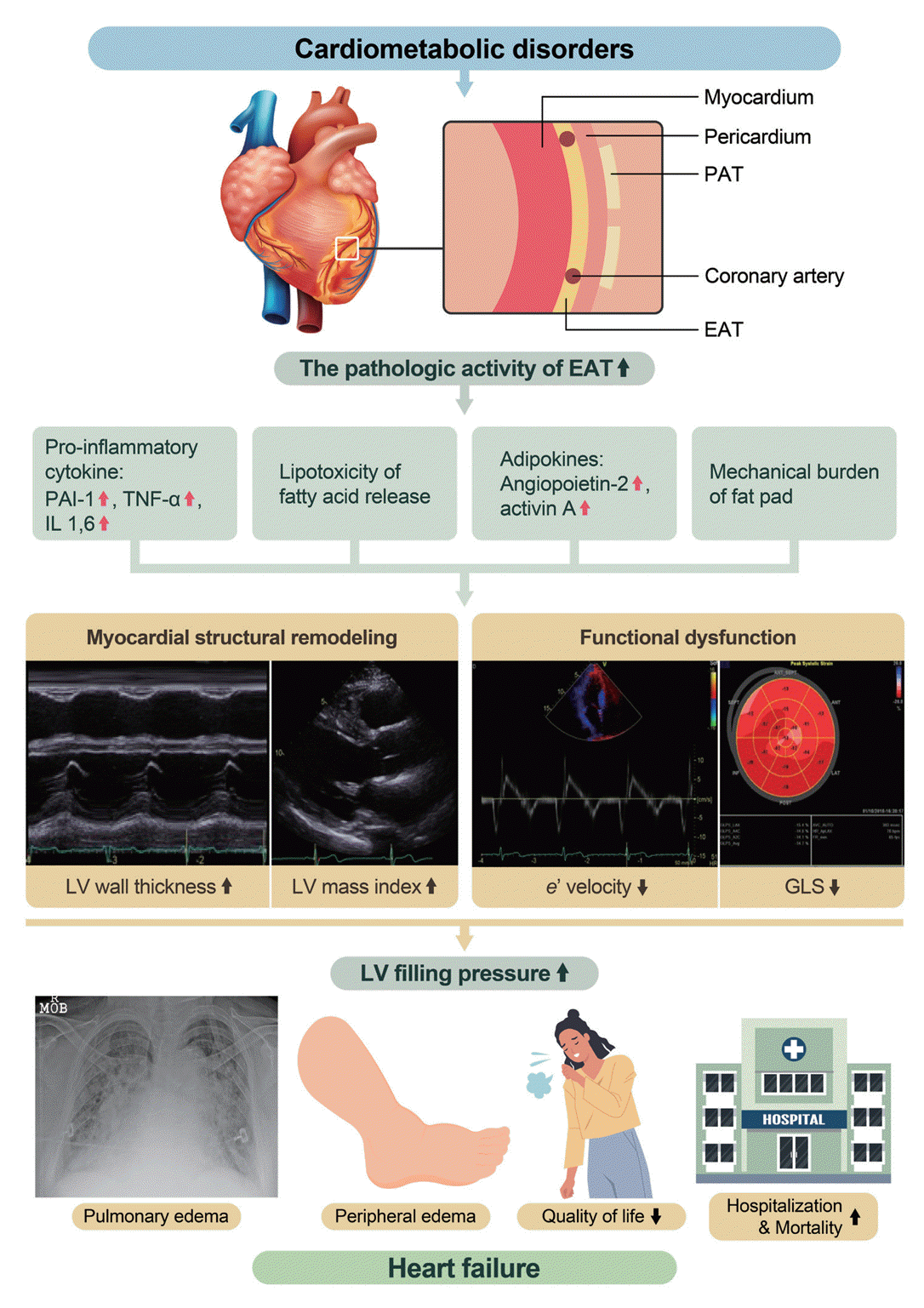
PATHOPHYSIOLOGIC MECHANISM OF EAT INDUCING MYOCARDIAL DAMAGE
Metabolic syndrome is a constellation of various cardiovascular risk factors, including hypertension, abdominal obesity, dyslipidemia, and insulin resistance [26]. Although body mass index (BMI) is commonly used as a marker of general obesity, it cannot accurately reflect the burden of visceral obesity because it does not differentiate between fat content and muscle mass [27,28]. Abdominal obesity, as assessed by measuring waist circumference (WC), is a well-established risk factor for myocardial dysfunction and development of CVDs [29,30]. For a more detailed evaluation, the amount of VAT can be quantified using bioelectrical impedance analysis. We have previously reported that VAT, as evaluated by bioelectrical impedance analysis, but not abdominal obesity evaluated by measuring WC, is correlated with myocardial structural and functional remodeling in individuals with early dysmetabolic states [17]. VAT secretes pro-inflammatory and pro-atherogenic cytokines that have adverse systemic effects on the cardiovascular system [31]. Moreover, VAT is an active source of several adipokines. In pathological conditions, dysregulated secretion of adiponectin and leptin contributes to changes in myocardial fatty acid metabolism, resulting in myocardial remodeling and dysfunction [32].
EAT, a component of VAT located in the heart, has metabolic, mechanical, and thermogenic roles. The heart is a vital organ with a high energy demand and no energy reserves in the myocardium. FFA oxidation through β-oxidation is a major source of energy, accounting for approximately 40% to 60% [33]. EAT has the ability to release and uptake FFAs, providing energy to the heart. Additionally, EAT acts as a buffer to protect the myocardium from exposure to toxic levels of FFA [34]. EAT also surrounds the epicardial coronary arteries, acting as a cushion, protecting against mechanical torsion [35]. EAT has a brown adipose tissue-like function that supplies heat to the myocardium [36].
In pathological conditions, EAT has unique effects on the myocardium compared to other VATs. Studies have investigated the inflammatory role of EAT and showed that CD8-positive T-cells and macrophage infiltration in EAT are highly increased in patients with CAD compared with those without CAD [37]. Furthermore, the level of serine proteinase inhibitor A3, which regulates the inflammatory status, was significantly increased in the EAT of patients with HF compared to those without HF [38]. In a study comparing EAT and subcutaneous adipose tissue (SAT) samples in patients with HF, p53 mRNA levels were highly up-regulated in EAT compared to those in SAT [39]. Chronic activation of p53 leads to the production of reactive oxygen species, adipose tissue apoptosis, and tissue inflammation [39]. In another study using EAT and SAT samples, p53 mRNA, which causes inflammation, was found to accelerate coronary atherosclerosis and myocardial remodeling. Notably, the inflammatory activity of EAT is not limited to local effects but also has systemic effects. EAT was found to be linearly related to high-sensitivity C-reactive protein (hs-CRP) levels in asymptomatic individuals who underwent a cardiovascular health survey [40]. Additionally, EAT thickness was reported to be linearly associated with hs-CRP levels in middle-aged men with suspected metabolic syndrome [41].
In addition to its inflammatory role, EAT is also involved in the regulation of myocardial energy metabolism. EAT stores FFAs and directly supplies them to the myocardium as an energy source. However, in pathological conditions, excessive release of fatty acids from EAT can lead to lipid infiltration into the myocardium, because EAT and the myocardium share the same coronary microcirculation and have no fascia separating them [42]. Several studies have reported an association between EAT volume and myocardial fat content [43,44]. For instance, a cardiac MRI study of healthy participants showed that increased EAT volume was independently associated with higher myocardial triglyceride content and worse left ventricular contractile function, even after controlling for visceral adiposity and other covariates [45]. This study also found an association between EAT volume and myocardial interstitial fibrosis, as assessed by T1 mapping. Another study showed that increased myocardial fat content was correlated with left ventricular diastolic dysfunction, particularly in patients with HF with preserved ejection fraction (HFpEF), indicating the potential role of EAT in the pathogenesis of HFpEF [46]. In addition to direct lipid infiltration, EAT has been implicated in the development of myocardial dysfunction through the dysregulated secretion of adipokines. Experimental studies using EAT samples have shown that EAT secretes adipokines, such as angiopoietin-2 and activin A, which can induce cardiomyocyte contractile dysfunction and altered cytosolic calcium fluxes in a dose-dependent manner [47].
EAT is a large fat pad that surrounds the myocardium. This protects the coronary arteries within EAT against torsion. However, excessive amounts of EAT can cause a mechanical burden on the myocardium, leading to myocardial dysfunction. This negative relationship between EAT and myocardial function has been observed in elderly women, with a stronger association in the lateral e´ and s´ than in the septal e´ and s´ [48]. This supports the hypothesis that the mechanical burden of EAT induces myocardial dysfunction.
In addition to its mechanical effects, EAT also plays an important role in the pathogenesis of coronary atherosclerosis. EAT surrounds the epicardial coronary artery and shares microcirculation. Inflammatory cytokines, adipokines, and metabolic substrates secreted from EAT affect the pathogenesis of coronary atherosclerosis [49]. EAT is not equally distributed throughout the heart and tends to accumulate excessively at the focal site. Thus, EAT appears to be a transducer of metabolic disturbances and systemic inflammation in the underlying coronary artery [42]. In patients with suspected CAD, EAT thickness is independently associated with the presence of obstructive CAD and vasospasm, as confirmed by invasive angiography [50]. Moreover, studies using CT have demonstrated that an increased EAT volume is related to the vulnerable type of plaque, suggesting that EAT may contribute to the progression and vulnerability of coronary plaque [51]. Interestingly, EAT has also been implicated in coronary microvascular dysfunction, which plays an important role in the development of HF without obstructive CAD [52]. In patients without obstructive CAD, increased EAT volume is related to microvascular dysfunction, as evaluated using Rb-82 positron emission tomography [53]. These findings suggest that EAT is involved not only in the pathogenesis of epicardial CAD, but also in coronary microcirculation dysfunction. Fig. 3 summarizes the pathophysiologic mechanism of EAT.
EAT AND THE RISK OF HF
The association between the amount of EAT and myocardial dysfunction has been investigated in various clinical situations, including the general population, patients with metabolic disease without CVD, and patients with established CVD. We previously reported that a greater EAT thickness was correlated with a higher left ventricle (LV) mass index, worse LV systolic dysfunction represented by LV global longitudinal strain, and LV diastolic dysfunction in patients with suspected metabolic syndrome and no overt CVD [1]. In elderly women without established CVD, an increase in the EAT amount was related to worse LV systolic and diastolic dysfunction [48]. In patients with acute myocardial infarction, EAT amount progressively increased according to the grade of LV diastolic dysfunction [54].
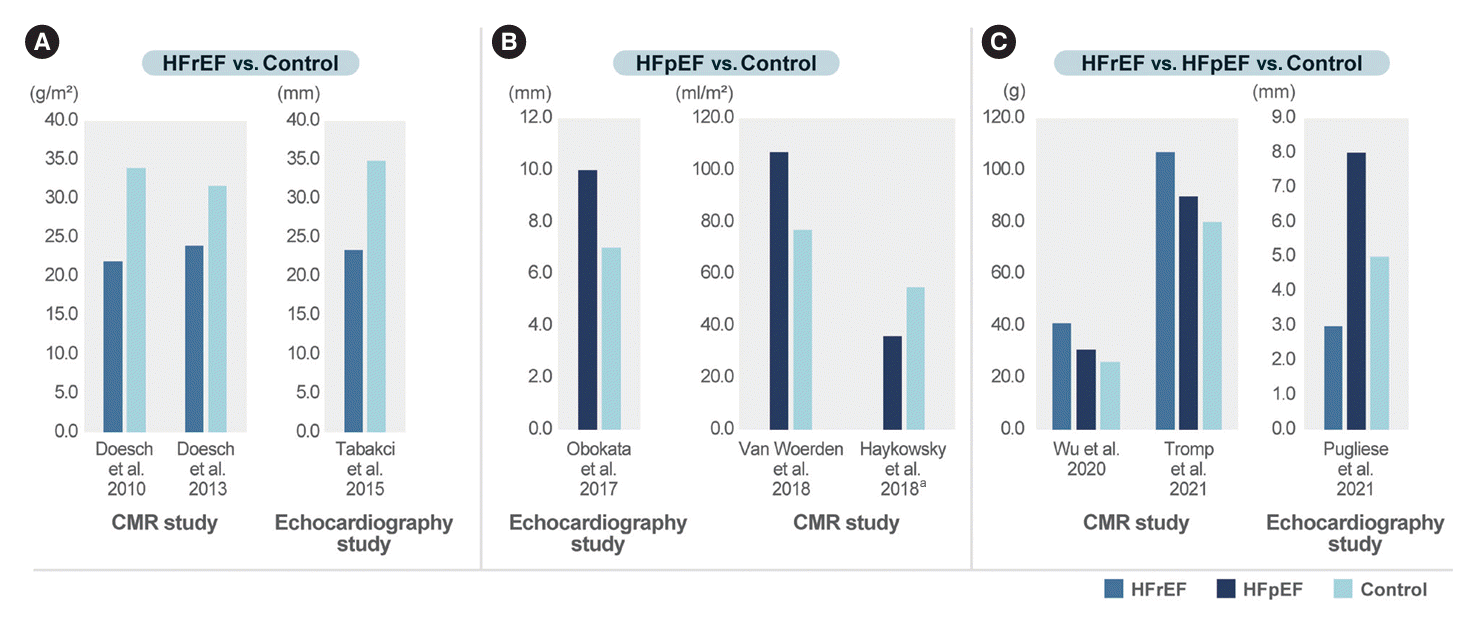
Excessive epicardial fat pad has an adverse effect on the myocardium. The amount of EAT varies according to the HF subtype: HF with reduced ejection fraction (HFrEF) or HFpEF. Fig. 4 shows the EAT amount in the HFrEF, HFpEF, and control groups. When patients with HFrEF and controls were compared, both cardiac MRI and echocardiography showed that the EAT amount was consistently reduced in patients with HFrEF than in controls [55-57]. However, the EAT amount in the HFpEF group was reported to increase in two studies and decrease in one study, when compared with that in the control group [58-60]. As the HFpEF study population was heterogeneous, this might have led to conflicting results. Few studies have compared patients with HFrEF and HFpEF with controls. In the study by Tromp et al. [61], which utilized both cardiac magnetic resonance (CMR) and echocardiography, it was observed that EAT thickness, as measured by echocardiography, was lower in HFrEF compared to the control group. However, EAT mass, as determined by CMR, was higher in the HFrEF group. It’s worth noting that the method of measuring EAT can significantly influence the study’s findings. Echocardiography primarily assesses EAT thickness at the RV free wall, whereas CMR provides a more comprehensive evaluation of EAT mass across the entire heart. Given that EAT covers a larger portion of the heart in cases of larger heart mass, Tromp et al. [61] indexed EAT mass to heart mass, revealing that the indexed EAT mass was lower in the HFrEF group when compared to the control. Thus, for CMR studies, it is necessary to measure EAT mass and adjust for body surface area.
Although the exact explanation for different status of EAT according to HF classification is not clear, we can propose some possible explanations. Studies with HFrEF mainly enrolled patients with ischemic cardiomyopathy and dilated cardiomyopathy. These advanced HF conditions not only leads to LV dysfunction but also RV dysfunction, followed by intestinal congestion. Intestinal congestion in HF can lead to anorexia and poor nutrition absorption, resulting in catabolic conditions such as sarcopenia [62]. This might be related to the decreased volume of EAT in HFrEF. Conversely, HFpEF is frequently accompanied by obesity, and this induces an increase in intravascular volume, which imposes hemodynamic stress by increasing myocardial workload [30]. Given the higher proportion of obese patients in HFpEF, EAT volume may increase in HFpEF. However, this issue requires further precise research focused on the classification of HFrEF and HFpEF. Pugliese et al. [63] conducted a more advanced study analyzing EAT thickness in patients with HFrEF, HFpEF, and controls and investigated the association between EAT thickness and multiple biomarkers and cardiorespiratory fitness evaluated by peak oxygen consumption. Increased EAT thickness was associated with worse cardiorespiratory fitness, biomarker profile, RV-pulmonary arterial uncoupling, and mortality in patients with HFpEF. Conversely, reduced EAT thickness was associated with worse LV dysfunction and mortality in patients with HFrEF. This study showed that EAT had a pathophysiological role in the characterization of HFrEF and HFpEF. Because EAT has a brown adipose tissue-like function, it serves as an energy reservoir and reflects the catabolic status of HFrEF. In HFpEF, EAT is associated with a worse biomarker profile, suggesting enhanced EAT activity. In HFpEF, EAT maybe associated with hemodynamic stress and poor cardiorespiratory fitness, leading to increased adverse events. In the study with HF with midly reduced ejection fraction and HFpEF, EAT accumulation was associated with the adverse prognosis after adjusting for the conventional risk factors and the severity of HF [64].
Contrary to the findings of various studies on EAT and prevalent HF, the implications of EAT burden on the development of HF have rarely been investigated. The Multi-Ethnic Study of Atherosclerosis group explored whether the baseline pericardial fat volume, including epicardial and pericardial fat, was linked to the occurrence of HF [65]. In this study, pericardial fat volume was evaluated in 6,785 community-based individuals by CT. Pericardial fat volume was linearly associated with an increased risk of HF after adjustment for baseline characteristics, including anthropometric parameters (1-standard deviation increase in pericardial fat volume: hazard ratio, 1.22; 95% confidence interval, 1.12 to 1.31; P<0.001). In the Jackson Heart study with 2,882 participants without prevalent HF, a higher volume of PAT and VAT was associated with an increased risk of HFpEF. Although the replication of these findings is warranted in other community-based studies, this study suggested that increasing the amount of EAT in a community-based population might be a novel risk factor for newly diagnosed HF.
EAT AS A POTENTIAL FOR THERAPEUTIC TARGET
EAT can be visualized and quantified using two-dimensional echocardiography or other imaging modalities. EAT volume changes more quickly than other anthropometric parameters of body fat [66]. The role of EAT as a therapeutic target has been elucidated using several emerging cardiovascular drugs. A randomized controlled trial (RCT) evaluated the effect of an intensive dose of atorvastatin and a moderate dose of pravastatin on the progression of the coronary calcium score evaluated by CT [67]. The investigators also evaluated the effect of statins on changes in the EAT volume. Only the intensive-intensity statin group showed significantly decreased EAT volume but not the moderate-intensity group. The degree of lipid reduction did not correlate with EAT regression. This finding suggested that the effect of statins on EAT volume might be related to pleiotropic effects, such as anti-inflammatory effects, and not lipid-lowering effects. In patients with aortic stenosis, statin treatment was associated with lower EAT thickness and an in vitro statin-modulated inflammatory profile of human EAT [68]. In patients with coronary artery stenosis, the use of 20-mg atorvastatin was associated with lower EAT thickness than that with the use of 10-mg simvastatin and ezetimibe [69]. However, the effects of statins on EAT thickness in patients with HF require further investigation.
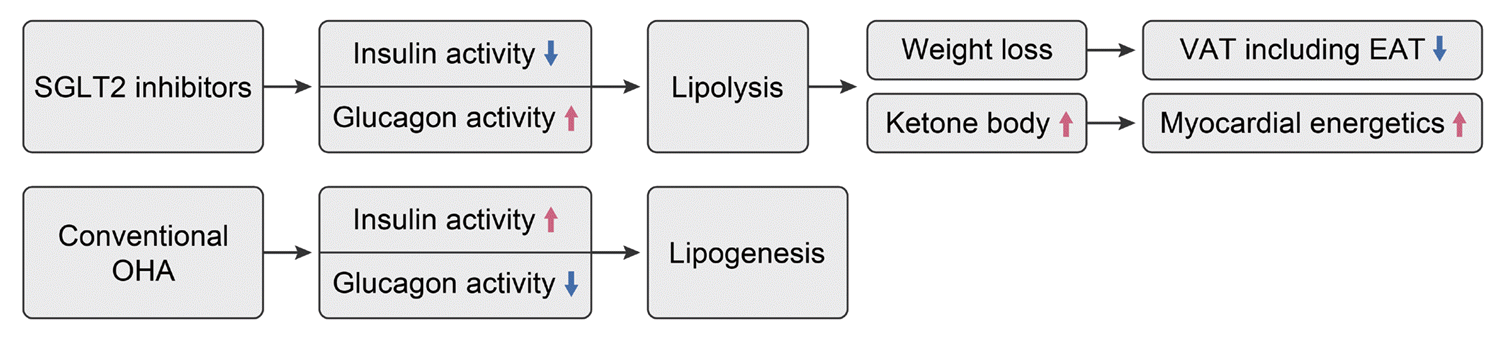
The lipolytic effect of sodium glucose cotransporter 2 (SGLT2) inhibitors, known as the statins of the 21st century [70], has been investigated with a focus on VAT, including EAT [71]. SGLT2 inhibitors increase glucose excretion in the urine, leading to reduced blood glucose levels. Consequently, insulin secretion decreases, and the release of glucagon, which has counter-regulatory effects, increases. Unlike insulin, which promotes lipogenesis and lowers blood glucose, glucagon stimulates lipolysis, elevating blood glucose levels. In other words, SGLT2 inhibitors promote the lipolysis through increased glucagon secretion, as evidenced by several studies showing that the use of SGLT2 inhibitors can reduce visceral obesity [72,73]. Lipolysis results in an increase in blood ketone levels, which can explain one of the notable side effects of SGLT2 inhibitors, known as ketoacidosis. Interestingly, blood ketones serve as an efficient metabolic energy source for the heart. Given the heart’s continuous need for hemodynamic energy, it relies on various substances as energy sources, making it a ‘metabolic omnivore.’ Since ketone produce highest adenosine triphosphate via ketone oxidation, ketone is often referred to as ‘the superfuel of the heart.’ This rise in blood ketone levels enhances the energy efficiency of the heart, leading to an improved prognosis for HF. In a non-diabetic pig model, use of SGLT2 inhibitor improved myocardial energetics and increased the myocardial ketone uptake [74]. This may explain the outstanding findings of improved HF-related outcomes with SGLT2 inhibitors (Fig. 5) [75]. Based on these findings, the effect of SGLT2 inhibitors on lowering EAT volume and activity has been studied in several studies, including RCTs [72,76-81]. Table 2 summarizes the major RCTs that have investigated the effects of SGLT2 inhibitors on EAT volume. The Iacobellis group studied the effect of dapagliflozin compared to placebo in patients with type 2 diabetes mellitus with a BMI ≥27 kg/m2 [79]. Dapagliflozin reduced EAT thickness by 20% from baseline in 6 months. The reduction in EAT thickness was higher in the dapagliflozin group than in the metformin group. In this study, the reduction in EAT thickness did not correlate with weight loss, suggesting that the EAT reduction effect of SGLT2 inhibitors might be mediated beyond weight loss. Requena-Ibanez et al. [81] performed an RCT comparing empagliflozin and placebo in patients with HFrEF without diabetes. Empagliflozin reduced the EAT volume compared to the placebo, as evaluated by cardiac MRI after 6 months. EAT reduction was associated with improvements in inflammatory biomarkers, myocardial fibrosis, and aortic stiffness. The lipolytic effect of SGLT2 inhibitors on EAT volume varies according to the study population, drugs, study duration, and imaging modalities. A few studies have failed to demonstrate the effects of SGLT2 inhibitors on EAT volume [77,78]. These studies enrolled a small-sized population with a low cardiovascular risk, and the study period was short. A meta-analysis of RCTs concluded that SGLT2 inhibitors significantly reduced EAT volume in patients with type 2 diabetes mellitus [80]. The effect of SGLT2 inhibitors on the amount of EAT and whether the intervention-induced reduction of EAT volume or activity leads to the improvement in clinical outcomes of HF remains unclear. Therefore, further studies are required to address this issue.
A recent study demonstrated that glucagon-like peptide-1 receptor agonists (GLP-1RA) improved quality of life in patients with HFpEF and obesity [82]. GLP-1RA also exhibit pleiotropic effects, such as weight loss and cardiovascular protection beyond glucose control. They reduce appetite, delay gastric emptying, and can even alter body fat distribution [72]. EAT expresses receptors for GLP-1, and GLP-1RA have been found to be associated with increased gene expression related to the differentiation of white to brown adipose tissue and decreased expression of pro-adipogenic genes [83]. In light of this background, researchers have explored the effects of GLP-1RA injection on EAT. In patients with type 2 diabetes mellitus and obesity, a 24-week liraglutide treatment successfully reduced echocardiographic EAT thickness from 9.6±2.0 to 6.2±1.5 mm, marking a 36% reduction [84]. Long-acting GLP-1RA, such as semaglutide, also decreased EAT thickness in a dose-dependent manner [85]. In a study involving liraglutide, the reduction in EAT thickness was independently correlated with LV mass reduction [84]. This suggests that the reduction of EAT thickness by GLP-1RA could serve as a surrogate marker for myocardial reverse remodeling. Further studies are needed to investigate the association between EAT reduction and cardiovascular protection.
CONCLUSIONS
EAT is a promising biomarker for identifying the HF phenotype and guiding treatment strategies because of its close anatomical and functional connection with the myocardium. It is easily measurable and modifiable, which makes it an attractive therapeutic target. As contemporary medicine for HF emphasizes individualized approaches based on HF phenotypes, further studies are required to fully explore the therapeutic potential of EAT.
 XML Download
XML Download