

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Stroke, a type of cerebrovascular disease, is the leading cause of disability worldwide [1]. Stroke can be divided into hemorrhagic stroke and ischemic stroke [2]. Cerebral ischemia caused by insufficient blood supply to the brain accounts for 80%–85% of all strokes [3]. Various mechanisms, including inflammation, oxidative stress, and disruption of blood-brain barrier (BBB), are associated with ischemic stroke [4]. Intravenous thrombolysis with recombinant tissue plasminogen activator (tPA) is commonly used in the treatment of ischemic stroke [5]. Unfortunately, the tPA therapy has a limited therapeutic window with severe adverse reactions, including cerebral ischemia/reperfusion injury and hemorrhagic transformation [6,7]. Clinically, acute cerebral ischemia/reperfusion injury may occur during the shock, cardiac arrest, or perioperative period for neurosurgery and cardiovascular surgery. Perioperative ischemia/reperfusion-induced brain injury is usually difficult to be monitored in time and contributes to poor prognosis. Therefore, it is needed to develop therapeutic options for the prevention of ischemia/reperfusion-induced brain injury [8].
BBB is a continuous endothelial membrane within brain microvessels; it protects the central nervous system from harmful factors and plays a key role in maintaining the homeostasis of the brain microenvironment [9]. BBB disruption during ischemic stroke leads to vasogenic edema and hemorrhagic transformation [10]. The formation of BBB tight junction involves transmembrane proteins, including claudins, occludin, tricellulin, and junctional adhesion molecules, which are linked to cytoskeletal filaments by interaction with accessory proteins, including zonula occludens (ZO) protein [11]. Matrix metalloproteinase-9 (MMP-9), a zinc-binding proteolytic enzyme, can degrade tight junctions and disrupt BBB integrity after ischemic stroke [12]. Thus, therapies designed to maintain BBB integrity are promising strategies for the treatment ischemia/reperfusion-induced brain injury.
Microglia are resident immune cells in the central nervous system and can maintain tissue homeostasis [13]. Under immunological stimuli, microglia transform from resting microglia to activated microglia [14]. Activated microglia secret inflammatory cytokines and contribute to neuroinflammation and neurodegeneration [15]. As reported, microglia can develop into classically-activated (proinflammatory, M1) and alternatively-activated (anti-inflammatory, M2) phenotypes [16]. After stroke, M1-type microglia secret proinflammatory molecules, including inducible nitric oxide synthase (iNOS), tumor necrosis factor-α (TNF-α), and interleukin (IL)-1β, which result in BBB damage, whereas M2-type microglia attenuate inflammation and damage to BBB [17]. Therefore, promoting microglia transforming from M1 to M2 phenotypes may be beneficial to attenuate neuroinflammation and BBB damage after ischemia/reperfusion-induced brain injury.
Mitogen-activated protein kinases (MAPKs) can mediate cell proliferation, differentiation, transformation, and survival [18]. p38 MAPK activated by the MAPK kinases 3 and 6 has contradictory functions in the pathogenesis of cerebral ischemia/reperfusion injury. Some reports show that activated p38 MAPK induces cerebral infarction by augmenting inflammation [19,20]. Opposingly, it has been shown that activation of p38 MAPK protects brain endothelial cells from apoptosis after stroke [21,22]. Collectively, the biological functions of p38 MAPK in ischemia remain controversial. Therefore, we validated the biological functions of p38 MAPK in cerebral ischemia/reperfusion injury using a p38 MAPK inhibitor.
Many studies have reported the neuroprotective effects of drugs in ischemia/reperfusion-induced brain injury. Wang et al. [23] reported that a single dose administration of imatinib (20 and 40 mg/kg) attenuated ischemia-reperfusion-induced behavioral deficits. Shin et al. [24] found that stiripentol treatment after transient ischemia ameliorated transient ischemia-induced cognitive impairment and BBB leakage in gerbils. Dexmedetomidine is a selective α2-adrenergic agonist with central antisympathetic and analgesic functions [25]. This compound has been reported to ameliorate BBB permeability in ischemic stroke [26,27], sepsis [28], and subarachnoid hemorrhage [29]. Additionally, dexmedetomidine has been shown to inhibit neuroinflammation by increasing microglial M2 polarization [30]. In the intestinal, lung, kidney, and renal ischemia/reperfusion injuries, dexmedetomidine can inactivate p38 MAPK signaling pathway [31-34]. Moreover, dexmedetomidine has been demonstrated to be effective in protecting against ischemia/reperfusion-induced brain injury [35,36]. However, whether dexmedetomidine improves BBB disruption and M1/M2 polarization via the MAPK pathways has not been investigated.
In this study, we established a rat model of cerebral ischemia/reperfusion injury by middle cerebral artery occlusion (MCAO) and reperfusion. We hypothesized that dexmedetomidine ameliorates ischemia/reperfusion-induced brain injury in rats by attenuating BBB permeability and neuroinflammation, downregulating MMP-9 levels, promoting M2 polarization, and inhibiting the MAPK pathways. This information might provide compelling evidence for the therapeutic effects of dexmedetomidine on BBB function in ischemic stroke.
Go to : 
METHODS
Ethical statement
All animal experiments were performed in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and were approved by the Ethical Review System for Laboratory Animal Welfare of the Wuhan Myhalic Biotechnology Co., Ltd (approval number: DWLL202106084).
Animal experiment
Male Sprague-Dawley rats (Vital River Co. Ltd.) weighing 180–200 g were housed under standard conditions (22°C ± 2°C, 55% humidity) with a 12/12 h light/dark cycle. All rats were given free access to food and water. Animals were operated with slightly modified MCAO model as described by Longa et al. [37]. Anesthesia was induced by 4% isoflurane and maintained by 2% isoflurane during MCAO operation. A midline neck incision was made to expose the left common carotid artery (CCA), internal carotid artery (ICA) and external carotid artery (ECA). After ligation of the distal trunk of ECA, the CCA and ICA were temporarily clamped using microvascular clips. A small nick was made in the ECA and a nylon monofilament (diameter: 0.28 mm) with a silicone tip (diameter: 0.37 ± 0.02 mm) (Sunbio Biotech) was inserted from the nick into the ICA. A laser Doppler flowmeter (PeriFlux5000; Perimed Co.) was used to monitor the regional cerebral blood flow. A value less than 20% indicated a successful occlusion. The filament was withdrawn after 1.5 h to allow reperfusion. After suturing the wound, the rats were returned to cages for recovery. During operation, the rat’s body temperature was maintained at 37°C ± 0.5°C using a heating pad. The sham operation underwent MCAO without monofilament insertion.
Grouping and drug administration
Rats were randomly divided into seven groups: sham; MCAO; MCAO + dexmedetomidine (Hengrui Pharmaceutical Co., Ltd.) (injected intravenously with 9 μg/kg dexmedetomidine 30 min after MCAO) [38,39]; MCAO + SB203580 (Shanghai Baililai Biotech) (injected intraperitoneally with 200 μg/kg SB203580 30 min before MCAO) [40,41]; MCAO + dexmedetomidine + anisomycin (Beyotime) (injected intravenously with 9 μg/kg dexmedetomidine plus intraperitoneally with 100 μg/kg anisomycin 30 min after MCAO) [42,43]; MCAO + dexmedetomidine + P79350 (Acmec) (injected intravenously with 9 μg/kg dexmedetomidine plus intraperitoneally with 100 μg/kg P79350 30 min after MCAO) [44]; MCAO + dexmedetomidine + EGF (Peprotech) (injected intravenously with 9 μg/kg dexmedetomidine plus intraperitoneally with 250 μg/kg EGF 30 min after MCAO) [45].
Evaluation of neurological function
Twenty-four hours after reperfusion, neurological deficits were evaluated using the Longa score as previously reported [46]. The scores are: 0 point: normal; 1 point: the forelimb contralateral to the lesion cannot be fully extended when pulling the tail; 2 points: turning to the opposite side of the lesion while walking; 3 points: falling to the contralateral side of the lesion when walking; 4 points: poor consciousness and falling to spontaneously walk.
Infarct volume
The infarct volume was measured as previously described [47]. After reperfusion, the rats were anesthetized and rapidly decapitated. The collected brains were cut into 2-mm-thick coronal slices. Then, the sections were stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC) (Sigma-Aldrich) for 10 min at 37°C and immersed in 10% formalin overnight. The infarction area was demarcated and analyzed using ImageJ software. The percent of the infarct volume was obtained by dividing the infarct volume by total volume of all the slices.
Histological changes
Brain tissues were fixed in 4% paraformaldehyde, dehydrated and paraffin-embedded, and cut into 5-μm-thick sections for hematoxylin-eosin (H&E) staining and Nissl staining as instructed by the manufacturer. Images of five randomly selected fields of each section were obtained using a phase contrast microscope (Olympus).
Evans blue (EB) staining
To assess BBB integrity, the rats were injected with 2% EB (2 ml/kg) through the caudal vein. After 1 h of EB injection, the brains were perfused with phosphate buffered saline (PBS) to clear EB from blood stream. Then, the rats were decapitated. The left hemisphere of the brain was weighed, placed in a tube containing 3 ml dimethylformamide (Sigma-Aldrich), and thawed in water at 60°C in the dark for 24 h. A microplate spectrophotometer was used to measure the optical density value of EB at 630 nm. The EB content was calculated from a standard EB curve.
Brain water content
After anesthesia, the left hemisphere of the brain was immediately resected and weighed to obtain the wet weight. Subsequently, the brain was dried in an oven at 100°C for 24 h and weighed to obtain the dry weight. Brain water content = [(wet weight – dry weight) / wet weight] × 100% [48].
Western blotting
The left hemisphere of the brain was homogenized in pre-cooled RIPA lysis buffer (Sigma-Aldrich) with protease inhibitor (ApexBio Technology) followed by centrifugation at 12,000 ×g at 4°C for 10 min. A Bicinchoninic Acid Assay (BCA) protein assay kit (Yeasen) was used to assess protein concentration. Protein (40 μg) was boiled in loading buffer, separated by 10% SDS-PAGE gels, and transferred onto PVDF membranes. After blocking with 5% nonfat milk, the membranes were incubated overnight with the primary antibodies against ZO-1 (ab190085, 1:1,000; Abcam), claudin-5 (bs-10296R, 1:1,000; Bioss), ERK1/2 (ab17942, 1:1,000; Abcam), MMP-9 (ab76003, 1:5,000; Abcam), occludin (ab216327, 1:1,000; Abcam), AQP-4 (bs-1087R, 1:1,000; Bioss), p38 (ab170099, 1:1,000; Abcam), GAPDH (ab181602, 1:10,000; Abcam), phosphorylated JNK (ab47337, 1:1,000; Abcam), phosphorylated p38 (ab4822, 1:1,000; Abcam), phosphorylated ERK1/2 (ab201015, 1:1,000; Abcam), JNK (ab199380, 1:2,500; Abcam), iNOS (ab178945, 1:1,000; Abcam), CD86 (sc-28347, 1:1,000; Santa Cruz Biotechnology), Arg-1 (#93668, 1:1,000; Cell Signaling Technology) and CD206 (sc-58986, 1:1,000; Santa Cruz Biotechnology) at 4°C. Then the membranes were incubated with secondary antibodies for 1 h at room temperature. The protein bands were visualized using BeyoECL reagent (P0018S; Beyotime) and a GeneGnome XRQ Chemiluminescence Imaging System (GeneGnome XRQ; Syngene) exposure instrument. Gray intensity of the bands was calculated using ImageJ 1.8.0 software. Finally, the expression levels of the proteins were standardized by densitometric analysis to GAPDH levels.
Immunofluorescence
The isolated brain tissues were fixed in 4% paraformaldehyde for 4 h and then immersed in phosphate buffer containing 30% sucrose overnight. Then the tissues were embedded in optimal cutting temperature solution, frozen at –30°C, and coronally cut into 8-μm sections. The sections were then blocked with 10% goat serum for 2 h and incubated overnight at 4°C with the primary antibodies against claudin-5 (bs-1241R, 1:300; Bioss), ZO-1 (ab221547, 1:100; Abcam), and occludin (ab216327, 1:100; Abcam). After washing, the sections were incubated with the corresponding secondary antibodies for 30 min at 37°C. The double-labeled sections were subsequently counterstained with DAPI. Images were captured using a fluorescence microscope (Olympus). ImageJ was used to measure the fluorescence intensity of the proteins on brain sections that were positively stained. Five randomly selected visual fields per section were analyzed. All images were transformed to 8-bit type and a threshold was set. Then, a rectangular box of similar size where the fluorescence intensity was uniformed was traced by wand tool to measure values of the fluorescence intensity. Results are expressed as percentage control values.
Gelatin zymography
Gelatin zymography was used to measure MMP-9 activity. Briefly, the tissues from the left hemisphere of the brain were added to a lysis solution (1 mM sodium fluoride, 1 mM sodium orthovanadate, 0.5% protease inhibitor, and 1 mM phenylmethyl sulfonyl) without ethylenediaminetetraacetic acid and incubated on ice for 30 min. The mixtures were then sonicated using Sonoplus ultrasonic homogenizers (Bandelin Electronics) and incubated on ice for 30 min. After a 15-min centrifugation (16,200 ×g) at 4°C, the supernatant was transferred to a new microtube. Copper sulfate solution (Sigma-Aldrich) and BCA solution were used to quantify the total protein concentration of the supernatant. Subsequently, protein (13 μg) was dissolved in non-reducing sodium dodecyl sulfate (SDS) sample buffer and resolved on a 7.5% acrylamide gel containing 0.1% gelatin without boiling. After electrophoresis, the gels were soaked in washing buffer (2.5% Triton X-100, 5 mM calcium chloride, 1 μM zinc chloride, and 50 mM Tris-HCl, pH7.5 in distilled water) twice for 30 min to remove SDS from the gel and then rinsed with incubation buffer (1% Triton X-100, 5 mM calcium chloride, 1 μM zinc chloride, and 50 mM Tris-HCl, pH7.5 in distilled water) for 10 min at 37°C. Gels were then placed in fresh incubation buffer and incubated for 24 h at 37°C with agitation to induce gelatinase reaction. After incubation for 24 h, gels were stained with Coomassie blue (Sigma-Aldrich) for 1 h and rinsed with distilled water. Stained gels were then incubated with destaining buffer until clear white bands appeared. The white band of MMP-9 was digitized and quantified using the ImageJ software.
RT-qPCR
Total RNA was isolated using TRIzol reagent (Beyotime) and quantified by NanoDrop (Thermo Fisher Scientific). The reverse transcription of RNA into cDNA was performed using the BeyoRT first strand cDNA synthesis kit (Beyotime). Then, the cDNA was amplified by RT-qPCR in a reaction containing SYBR Green PCR Master Mix (Solarbio) and analyzed using the ABI 7500 Fast Real Time PCR System (Thermo Fisher Scientific). Data were normalized to GAPDH expression levels. The 2–△△Ct method was used to calculate the expression levels of mRNAs [49]. The primers used in this study are listed in Table 1.
Table 1
Sequences of primers used for reverse transcription-quantitative PCR
![]()
Enzyme-linked immunosorbent assay (ELISA)
After being homogenized in pre-cooled PBS, the brain tissues were exposed to freezing and thawing in liquid nitrogen, and the homogenates were centrifuged at 10,000 ×g at 4°C. TNF-α and IL-1β levels in the supernatant were evaluated using commercially available ELISA kits (Enzyme-linked Biotechnology) according to the manufacturer’s protocol.
Statistics analysis
Data was analyzed by GraphPad Prism 8.0 software and are expressed as the mean ± standard deviation. All data were analyzed by Shapiro–Wilk to evaluate whether they fitted a normal distribution. All data in this study conformed to a normal distribution (p > 0.1). p < 0.05 was considered statistically significant. Comparisons among multiple groups were obtained using one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc analysis.
Go to :
RESULTS
Dexmedetomidine ameliorates MCAO-induced brain injury and neurological deficits
Fig. 1A presented the flow chart of the experimental protocols. The results of TTC staining showed that MCAO induced a high percent of infarct volume. However, administration of dexmedetomidine or SB203580 reduced the infarct area induced by MCAO (Fig. 1B, C). H&E staining revealed that the sham group exhibited intact cellular structure and orderly tissue arrangement. However, disordered brain tissues, damaged neuronal structure, and necrosis were observed in MCAO rats. Notably, administration of dexmedetomidine and SB203580 attenuated the detrimental effects of MCAO on cellular structure and tissue arrangement (Fig. 1D). As Nissl staining revealed, MCAO led to shrunken cell bodies, pyknosis, and decreased number of Nissl bodies. Dexmedetomidine or SB203580 increased the number of Nissl bodies and ameliorated neuronal injury (Fig. 1E). Neurological function was examined using the Longa scoring method, and we found that MCAO induced significant neurological dysfunction, which was attenuated by dexmedetomidine or SB203580 (Fig. 1F).
 | Fig. 1Dexmedetomidine reduces infarct volume and neurological deficits.(A) Schematic of the experiment protocols. (B, C) Effects of dexmedetomidine or SB203580 on the infarct volume were detected by TTC staining. (D) Representative photomicrographs of H&E staining in the rat brain in the four groups. Scale bar = 100 μm. (E) Representative images of Nissl staining in the rat brain. Scale bar = 100 μm. (F) Effects of dexmedetomidine or SB203580 on the neurological deficit scores. N = 8. Results are presented as mean ± SD, and statistical significance was performed by one-way ANOVA. MCAO, middle cerebral artery occlusion; i.p., intraperitoneal; i.v., intravenous; TTC, 2,3,5-triphenyltetrazolium chloride. ***p < 0.001. ###p < 0.001.
|
Dexmedetomidine reduces BBB permeability in MCAO rats
To investigate the role of dexmedetomidine in BBB integrity and permeability, EB staining was conducted in the rat brain. The results demonstrated that dexmedetomidine or SB203580 reversed the MCAO-induced increase in EB extravasation (Fig. 2A). Additionally, MCAO increased brain water content, while administration of dexmedetomidine or SB203580 reduced brain water content in MCAO rats (Fig. 2B). As Western blotting revealed, MCAO decreased ZO-1, claudin-5, and occludin protein levels and increased AQP-4 protein levels in the brain. However, administration of dexmedetomidine or SB203580 restored the protein levels of ZO-1, claudin-5, occluding, and AQP-4 (Fig. 2C). As immunofluorescence showed, dexmedetomidine or SB203580 treatment restored the fluorescence intensity of ZO-1, claudin-5, and occludin reduced by MCAO in the brain, consistent with the Western blotting results (Fig. 2D).
 | Fig. 2Dexmedetomidine alleviates BBB disruption in MCAO rats.(A) BBB permeability was detected by EB staining in the brain. (B) Effects of dexmedetomidine or SB203580 on the brain water content. (C) Western blotting was performed to measure ZO-1, claudin-5, occluding, and AQP-4 protein levels. (D) Immunofluorescence staining of claudin-5, ZO-1, and occludin protein levels. Scale bar = 100 μm. N = 8. Results are presented as mean ± SD, and statistical significance was performed by one-way ANOVA. BBB, blood-brain barrier; MCAO, middle cerebral artery occlusion; EB, Evans blue. ***p < 0.001. ###p < 0.001.
|
Dexmedetomidine reduces MMP-9 levels in MCAO rats
Western blotting indicated that the protein levels of MMP-9 were upregulated after MCAO. However, administration of dexmedetomidine and SB203580 inhibited the MCAO-induced increase in MMP-9 protein levels (Fig. 3A). According to the results of gelatin zymography, the MMP-9 activity was elevated in MCAO rats, while administration of dexmedetomidine or SB203580 reversed the effects of MCAO on MMP-9 activity (Fig. 3B).
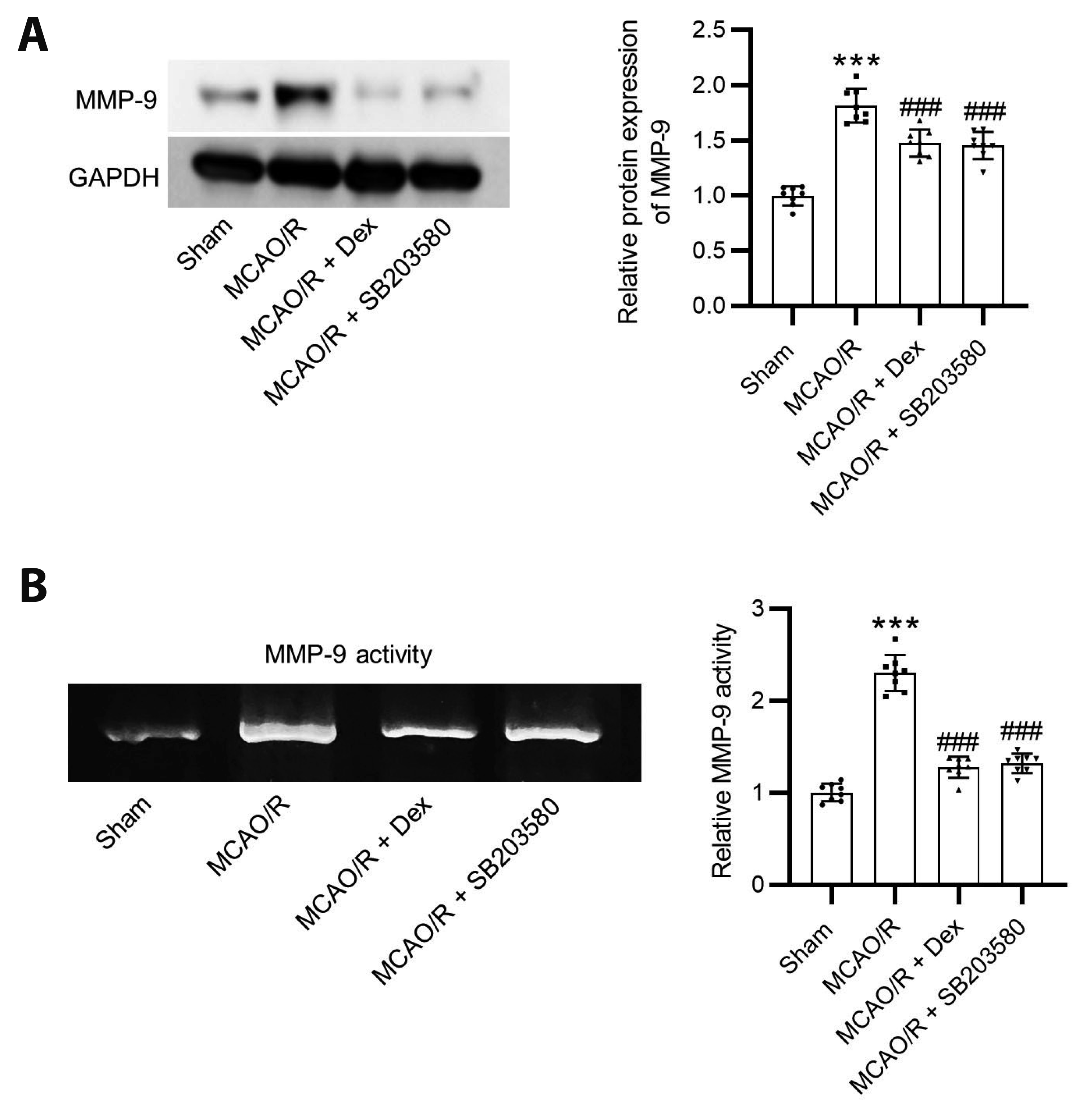 | Fig. 3Dexmedetomidine reduces MMP-9 levels in MCAO rats.(A) Western blotting of MMP-9 protein levels. (B) The MMP-9 activity was detected by Gelatin zymography. N = 8. Results are presented as mean ± SD, and statistical significance was performed by one-way ANOVA. MMP-9, matrix metalloproteinase-9; MCAO, middle cerebral artery occlusion. ***p < 0.001. ###p < 0.001.
|
Dexmedetomidine promotes M2-type microglial polarization
We employed RT-qPCR and Western blotting analyses to analyze the levels of M1-type markers (iNOS, CD86) and M2-type markers (CD206, Arg-1). The results revealed that the MCAO-induced significant increases in iNOS and CD86 expression levels were notably reversed by dexmedetomidine or SB203580. Additionally, CD206 and Arg-1 expression levels were further increased by dexmedetomidine or SB203580 in the ischemic hemisphere (Fig. 4A–F). Additionally, TNF-α and IL-1β levels were upregulated after MCAO, which were reduced by dexmedetomidine or SB203580 (Fig. 4G, H), suggesting that dexmedetomidine or SB203580 promoted M2-type microglial polarization to inhibit inflammatory response.
 | Fig. 4Dexmedetomidine promotes M2-type microglial polarization.(A-D) iNOS, CD86, CD206 and Arg-1 mRNA levels were measured by RT-qPCR. (E, F) iNOS, CD86, Arg-1 and CD206 protein levels were measured by Western blotting. (G, H) TNF-α and IL-1β levels were examined by ELISA. N = 8. Results are presented as mean ± SD, and statistical significance was performed by one-way ANOVA. iNOS, inducible nitric oxide synthase; TNF-α, tumor necrosis factor-α; IL, interleukin; MCAO, middle cerebral artery occlusion. ***p < 0.001. ###p < 0.001.
|
Dexmedetomidine inhibits MAPK pathways in MCAO rats
As Fig. 5A showed, MCAO significantly increased p38 phosphorylation, while dexmedetomidine or SB203580 attenuated the MCAO-induced increase in p38 MAPK phosphorylation. Additionally, we found that dexmedetomidine effectively reversed the MCAO-induced increases in ERK1/2 and JNK phosphorylation (Fig. 5B).
 | Fig. 5Dexmedetomidine or SB203580 reduces p38 MAPK phosphorylation.(A) The ratio of p-p38/p38 was evaluated by Western blotting. (B) The ratios of p-ERK1/2/ERK1/2 and p-JNK/JNK were assessed by Western blotting. N = 8. Results are presented as mean ± SD, and statistical significance was performed by one-way ANOVA. MCAO, middle cerebral artery occlusion. ***p < 0.001. ###p < 0.001.
|
Activation of JNK and p38 MAPK reverses the protective effects of dexmedetomidine in brain injury
Considering the inhibitory effects of dexmedetomidine on MAPK pathways, we investigated whether rescuing dexmedetomidine-associated reductions in p-p38, pERK1/2, and p-JNK can affect the neuroprotective effects of dexmedetomidine against ischemic brain injury. We used anisomycin (a JNK agonist), P79350 (a p38 agonist), and EGF (an ERK agonist) in MCAO rats. The results showed that dexmedetomidine significantly ameliorated increased infarct volume, EB extravasation, and brain water content induced by cerebral ischemia/reperfusion injury, and these effects were significantly reversed by administration of anisomycin or P79350 (Fig. 6). However, treatment with EGF did not reverse the effects of dexmedetomidine. These findings suggested that activation of JNK and p38 MAPK was responsible for the beneficial effects of dexmedetomidine in ischemic brain injury.
 | Fig. 6Activation of JNK and p38 MAPK reverses the protective effects of dexmedetomidine in brain injury.(A, B) Infarct volume was detected by TTC staining. (C) BBB permeability was detected by EB staining in the brain. (D) Brain water content in each group. N = 8. Results are presented as mean ± SD, and statistical significance was performed by one-way ANOVA. MCAO, middle cerebral artery occlusion; BBB, blood-brain barrier; EB, Evans blue. ***p < 0.001. ###p < 0.001. &&&p < 0.001.
|
Go to :
DISCUSSION
Reperfusion following cerebral ischemia leads to sudden recanalization of blood flow, causing additional ischemic injuries [50]. The MCAO model is widely used for research on ischemic brain injuries. This model has the advantages of easy operation, limited wounds, high reproducibility, and functions that produce brain edema with disrupted BBB [51]. Dexmedetomidine is a selective α2-adrenoceptor agonist and has been found to reduce neuronal injury and improve functional outcomes in multiple cell and animal models, including hypoxia-induced neuronal injury, ischemia/reperfusion injury, traumatic brain injury, intracerebral hemorrhage, epilepsy, neurodegeneration, and neuroinflammation [52]. A variety of mechanisms are associated with the neuroprotective effects of dexmedetomidine, including the inflammatory response, oxidative stress [53], autophagy, mitochondrial function [54], neurotransmitter regulation, and apoptotic pathway [55]. This study was the first to investigate the effects of dexmedetomidine on BBB function and M1/M2 microglial polarization via the MAPK pathways during the ischemia/reperfusion-induced brain injury.
BBB disruption contributes to cerebral edema and aggravates neuroinflammation [56]. Dexmedetomidine maintains BBB integrity in ischemia by inhibiting endothelial mitochondrial dysfunction [26]. Dexmedetomidine protects BBB integrity and alleviates brain edema in animal model of subarachnoid hemorrhage [29]. In mice with intracerebral hemorrhage, dexmedetomidine attenuates BBB permeability and alleviates the secondary cerebral injury [57]. In the current study, dexmedetomidine administration improved BBB integrity, as evidenced by decreased EB content. This finding was consistent with previous finding. The major tight junction-associated proteins of the BBB are occludin, claudins, and junctional adhesion molecules that are intracellularly linked to the adaptor protein ZO-1 [58]. AQP-4 is the most abundant water-permeable channel found in the mammalian brain. Cerebral injury may trigger AQP-4 upregulation and contribute to edema formation [59]. This study showed that dexmedetomidine treatment led to increase in the levels of tight junction proteins (ZO-1, claudin-5, and occludin) and decrease in the levels of AQP-4 in rats after cerebral ischemia/reperfusion injury, which has not been investigated previously. Additionally, the mechanisms dexmedetomidine ameliorating BBB disruption are largely unknown. MMPs are proteolytic enzymes that degrade the extracellular matrix [60]. MMP-9 is most closely associated with BBB damage after cerebral ischemia [60]. Knocking down MMP-9 gene upregulated the expression of tight junction proteins and decreases the permeability of BBB [61]. Activation of MMP-9 contributes to the early formation of vasogenic edema after transient cerebral ischemia and reperfusion [62]. Dexmedetomidine decreases serum concentration of MMP-9 in elderly patients with mild cognitive impairment, alleviating postoperative delirium [63]. Intrathecal preconditioning with dexmedetomidine suppresses spinal cord ischemia/reperfusion-induced increase in MMP-9 activity in rats [64]. Whether dexmedetomidine protects BBB integrity in cerebral ischemia/reperfusion injury by regulating MMP-9 has not been investigated. The current study showed that dexmedetomidine administration decreased MMP-9 activity and MMP-9 protein levels in brain tissues of MCAO rats. Thus, we concluded that dexmedetomidine ameliorated BBB disruption by inhibiting MMP-9.
In the acute phase of cerebral ischemia/reperfusion injury, neuroinflammatory cascades lead to BBB disruption, apoptosis, cerebral edema, and hemorrhagic transformation [65]. The proinflammation-dominant M1-type microglia produce proinflammatory cytokines and aggravate brain injury, whereas the antiinflammation-dominant M2-type microglia can clear tissue debris and promote neuroprotection [66,67]. In rats with cerebral ischemia/reperfusion injury, dexmedetomidine reduces the inflammatory response and autophagy effects by inhibiting the JNK pathway [68]. Dexmedetomidine reduces neuroinflammation in lipopolysaccharide-treated BV2 cells by reversing microglial M1/M2 polarization [30]. Dexmedetomidine ameliorates perioperative neurocognitive disorders via regulation of M1/M2 polarization [69]. Additionally, dexmedetomidine promotes M2-polarization of microglia and thus protects against ischemic brain injury [70]. However, whether dexmedetomidine promotes M2 microglial polarization after cerebral ischemia/reperfusion injury is unknown. In this study, we found that dexmedetomidine significantly decreased iNOS and CD86 levels and upregulated CD206 and Arg-1 levels, thereby altering microglial M1/M2 polarization.
The family members of MAPK, including p38 MAPK, ERK1/2, and JNK protein, are essential mediators or cellular growth, inflammation, and apoptosis [71]. SB203580 is widely used to inhibit p38 MAPK activation [72]. The association between dexmedetomidine and p38 MAPK in cerebral ischemia/reperfusion injury has not been studied. We found that the p38 MAPK inhibitor (SB203580) reduced the infarct volume, maintained the BBB integrity, and attenuated the inflammatory response in rats after cerebral ischemia/reperfusion injury. In this study, the protective effects were observed in both dexmedetomidine and SB203580. However, dexmedetomidine reduced p38, ERK, and JNK phosphorylation while SB203580 did not affect ERK and JNK phosphorylation. Dexmedetomidine can target various signal pathways as α2-adrenoceptor agonist, while SB203580 (p38 MAPK inhibitor) also shows inhibitory activity against GAK, CK1, RIP2, c-Raf, and GSK3 [73], which may cause different molecular mechanisms. To investigate whether the MAPK pathways were involved in the protection of dexmedetomidine in ischemic brain injury, we rescued the dexmedetomidine-associated reduction in p-p38, p-ERK1/2, and p-JNK using their agonists. We found that administration of anisomycin (JNK agonist) or P79350 (p38 agonist) aggravated infarct volume, EB extravasation, and brain water content in dexmedetomidine-treated rats. However, treatment with EGF did not reverse the effects of dexmedetomidine. These findings suggested that activation of JNK and p38 MAPK was responsible for the beneficial effects of dexmedetomidine in ischemia/reperfusion brain injury.
There are several limitations in this study. First, animal experiments were performed at only one point (24 h following MCAO). We did not set a series of time points to observe the changes of each experimental index. Second, only one dose of dexmedetomidine was administrated to rats, and multiple doses of dexmedetomidine are required to observe the efficacy of different doses of dexmedetomidine in ischemic brain injury. Third, the mechanisms dexmedetomidine inhibiting the phosphorylation of JNK and p38 are largely unknown. The resolution of these issues will help to understand the mechanisms of dexmedetomidine’s protective effects and provide important research foundations.
In conclusion, this study was the first to demonstrate that dexmedetomidine blocked the JNK and p38 MAPK signaling pathways after cerebral ischemia/reperfusion injury and thus ameliorated BBB permeability, neuroinflammation, MMP-9 activity, and promoted M2 microglial polarization. This study might provide a theoretical basis for the application of dexmedetomidine in the treatment of cerebral ischemia/reperfusion injury.
Go to :
 XML Download
XML Download