

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Cerebral cavernous malformation (CCM) is a vascular anomaly found in approximately 0.5% of population [2]. CCM is characterized by intermingled sinusoidal vessels without high-flow arteriovenous shunting. Typically, CCM lacks intervening brain parenchyma. In 2018, the International Society for the Study of Vascular Anomalies classified it as a form of venous malformations [13]. CCM differs from developmental venous anomaly (DVA), a common aberrant venous channel found in the deep subcortical area. However, the frequent co-occurrence of CCM and DVA supports the possibility of their shared origins [6]. Although other terms such as cavernous angioma or cavernoma have been used, “cavernous malformation” has become the standard, emphasizing its nature as a malformation rather than a neoplasm.
PEDIATRIC CCM
CCMs are typically diagnosed in young adults, with the peak age at diagnosis in the third and fourth decades of life [49]. However, around one third of patients with CCMs are diagnosed during pediatric ages [36], although solid statistics on this proportion are lacking due to biases in clinical studies. Previously, CCM was considered as a congenital lesion [1], with reports even suggesting in utero diagnosis [20]. However, CCM can develop postnatally, especially adjacent to preexisting DVA [11,14]. The discovery of causative genes for familial CCM and well-known association of radiation therapy and de novo genesis of CCMs also support the notion that CCM is a postnatal disease. Consistent with this, the prevalence of CCM increases with age, with rates of only 0.23% in infants, 0.56% in 6–12 years, and 0.88% in 13–18 years reported in one study [2].
Approximately 15–30% of patients have multiple CCMs [2,36]. In general, 80% of patients with CCMs have sporadic disease while the remaining 20% are considered familial disease [15]. However, more than half of patients with multiple CCMs have a family history, with reports indicating family history in up to 75% of these cases. The most frequent site of CCM occurrence is supratentorial, especially in subcortical areas. However, CCMs can develop in anywhere in the brain, including the basal ganglia, cerebellum, brainstem, and optic chiasm. A study on pediatric CCMs reported that supratentorial (lobar) locations constituted 78% of cases, followed by the brainstem (8%), thalamus and basal ganglia (7%), and cerebellum (6%) [19].
CLINICAL FEATURES
Clinical presentation
Intralesional bleeding is common in CCMs, and their volumes fluctuate over time, resulting in a slowly incremental slope. Occasionally, extralesional bleeding can occur, leading to acute expansion and mass effects on adjacent brain tissue [3]. Repeated hemorrhages and the absorption of blood products induce inflammation and leave a typical hemosiderin rim on magnetic resonance images (MRIs), along with tough gliotic layers surrounding the lesion. However, hemorrhage from CCMs is typically not life-threatening, unlike hemorrhages from arteriovenous malformations.
Common clinical presentations of pediatric patients with CCMs include headache, focal neurological deficits (FNDs), and seizures [49]. Headaches and FNDs are typically associated with acute or subacute bleeding, while seizures can result from both subacute bleeding and chronic inflammatory changes around the lesion. The occurrence of FNDs depends on the location of the CCM [2]. Lesions in the brainstem and deep cerebral regions such as the basal ganglia and thalamus frequently cause FNDs because minor hemorrhages and perilesional edema can affect critical nuclei or white matter tracts within these areas.
However, it’s important to note that 25–45% of pediatric patients with CCMs are asymptomatic, with the lesions being incidentally discovered [2,19,43]. The advancement of MRI technologies, especially the widespread use of susceptibility-weighted imaging (SWI) in routine brain imaging, has led to the increased detection of incidental CCMs in the pediatric population.
Natural history of pediatric CCMs
Understanding the natural history is crucial due to its benign nature and the fact that many affected individuals remain asymptomatic throughout their lives. The debate over hemorrhage rates in CCM has persisted, partly due to variations in definition of clinically meaningful hemorrhages. To enhance clarity, the term “symptomatic documented hemorrhage” is recommended, denoting acute or subacute symptoms (such as headache, FND, or seizures) along with radiologically, pathologically, or surgically confirmed intra- or extra-lesional hemorrhage [3]. Importantly, the definition does not require a simple increase in lesion size or the presence of a hemosiderin rim on imaging.
The reported hemorrhage rate of CCM in children and young adults was 1.6% per patient-year and 0.9% per lesionyear [2]. Intriguingly, the same study reported a much higher hemorrhage rate (8.0%) in symptomatic patients compared to incidental cases (0.2%). In adult studies, the 5-year cumulative rates of symptomatic hemorrhage are approximately 20% [4,22]. A comprehensive meta-analysis identified initial symptomatic presentation with bleeding and a brainstem location as significant risk factors of symptomatic hemorrhage during follow-up [22]. Particularly noteworthy is the high hemorrhage risk of patients with brainstem CCM presenting with intracerebral hemorrhage (ICH) or FND, estimated at 30.8% [22]. However, age, sex, and multiplicity of CCM were not identified as risk factor in the meta-analysis. Similar findings were observed in a cohort study of untreated pediatric CCM patients [43]. The 5-year cumulative risk of symptomatic hemorrhage was 15.9% in 129 patients aged ≤18 years. Higher cumulative risks were observed in patients with initial presentation of ICH (30.2%), those with a brainstem location (29.5%), and individuals with family history of CCM (17.2%). It remains uncertain whether brainstem CCM really bleeds more frequently or if symptomatic hemorrhages are over-represented in this location due to the brainstem’s vulnerability. Overall, one third of pediatric patients with brainstem CCM experience an aggressive clinical course characterized with recurrent hemorrhage and neurological decline [52].
Numerous studies on CCM have documented temporal clustering of hemorrhages, with a higher frequency of symptomatic hemorrhages occurring within 2 or 3 years following the initial hemorrhagic event [2,8,49]. Gross et al. [19], found that annual hemorrhage rate (AHR) for pediatric CCMs with a hemorrhagic presentation was highest at 18.2% in the first 3 years, decreasing to 4.8% after 3 years and further to 3.3% after 5 years. The temporal pattern is important for patient counseling and assessment of treatment efficacy [19].
Epilepsy associated with CCM
Seizures are the most common clinical symptom of CCM [47]. Moreover, CCM represents a significant structural etiology of chronic drug-resistant epilepsy. Data from the European Epilepsy Brain Bank (EEBB) which collects surgical specimens from epilepsy surgeries, revealed that vascular malformations constituted 6.1% of all specimens, ranking next only to hippocampal sclerosis, tumors, and cortical malformations [10]. Among vascular malformations, CCM was the most prevalent entity. Interestingly, in the EEBB data, the proportion of vascular malformations was 3.1% in children but rose to 7.2% in adults, reflecting an increasing incidence of CCM (and arteriovenous malformations) with ages.
CCMs lack functioning neural tissue within them, and are not inherently epileptogenic; however, they can trigger epileptic seizures by affecting adjacent brain tissue [7]. Extravasation of blood product and the resulting formation of a hemosiderin rim may contribute to epileptogesis [24,53]. Histopathological studies of perilesional tissues in CCM have shown a higher frequency of gliosis and reactive astrocytes in CCMs associated with epilepsy. Notably, siderotic vessels were found exclusively in perilesional tissues of CCMs associated with epilepsy [40].
The location of CCM is strongly correlated with development of epilepsy. One study demonstrated that 87% of the patients with temporal lobe CCM had epilepsy, with 50% of them experiencing drug-resistant epilepsy, whereas only 15% of those with non-temporal CCM had drug-resistant epilepsy [47]. The presence of a hemosiderin rim on imaging was identified as an independent risk factor, supporting its role in the epileptogenesis [55].
Infantile CCM
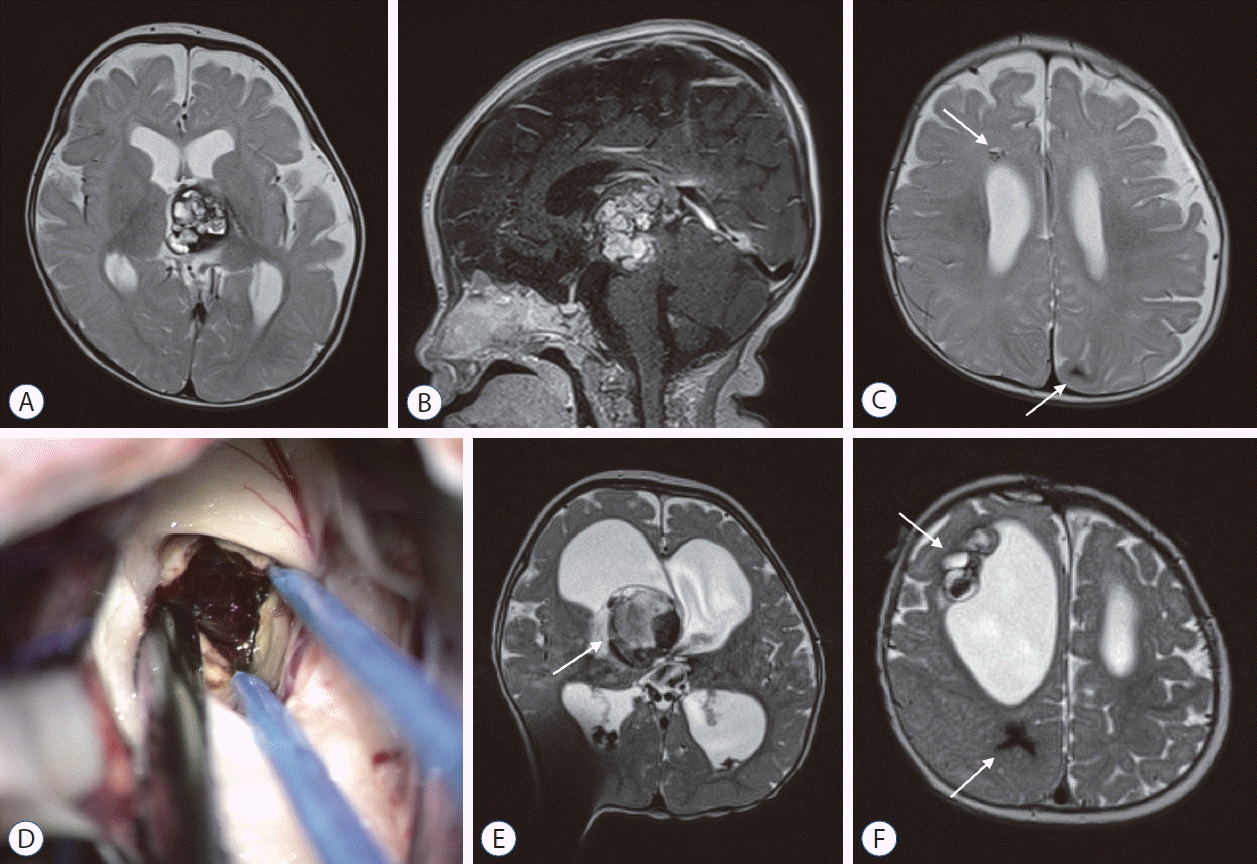
A small proportion of CCM develop during infancy (Fig. 1). In their review of data from 36 infantile patients with CCMs before the age of 2, Goyal et al. [18] found that nearly all but one patient presented with symptoms, predominantly seizures or FNDs. Notably, the proportion of CCMs located in the brainstem was higher (30%) compared to usual proportion of less than 10% in all-age cohorts. While thirteen patients were initially managed conservatively, no symptomatic hemorrhage was observed during 26 total person-years of observation. However, it is important to note that nearly two-thirds of patients in the literature review received surgery shortly after initial presentation, so the observed low hemorrhage rate during follow-up cannot be generalized. Some CCMs are detected prenatally through ultrasonography [7]. These congenital CCMs may lead to bleeding and symptoms in utero, potentially causing significant brain damage and fetal problems [20,21,28].
TREATMENT
Surgical treatment
Treatment of CCM is typically recommended when symptoms related to CCM develop and persist. Hemorrhage from a CCM can result in acute or subacute symptoms, such as headaches, although these symptoms often gradually subside in many patients. However, recurrent hemorrhages can lead to fluctuating symptoms or a stepwise decline in neurological functions due to incomplete recovery from FNDs, thereby justifying the need for definitive treatment. Seizures refractory to medical treatment is another major indication of treatment [36].
Surgical removal of CCM is the mainstay of treatments. Cortical and subcortical CCMs can be approached directly. Typically, surgery is performed through a small cortical incision, and sometime through a deep corridor to access the lesion. Removing liquefied portions of CCM containing subacute blood is advisable to reduce the brain pressure and create a working space for dissection. Many CCMs are surrounded by a gliotic layer, which serves as a good dissection plane during surgery. Complete removal of a CCM is crucial. A study revealed that out of 126 patients, 24 had a remnant CCM after surgery, and seven of them experienced at least one recurrent hemorrhage [16]. It is important to identify any DVA associated with CCM preoperatively and preserve it during surgery, as DVAs drain normal brain tissues nearby [39].
For patients with CCM-related epilepsy, the seizure-free rate after surgery is 75–85% [31,42]. One study on children presenting with CCM-related seizures revealed that the probability of anti-seizure medication discontinuation at 1 year is significantly higher in patients undergoing surgery (88%) compared to those under observation (53%) [48]. Several studies have reported that excision of the hemosiderin-stained rim along with CCM itself leads to better seizure outcome than removal of only the CCM, but these studies are small-sized and retrospective [9,23,50]. Moreover, some studies found no difference in outcome [12,54]. More data are needed to resolve this issue. A meta-analysis indicated that extended lesionectomy, involving cortical resection beyond the boundary of CCMs either with or without electrocorticography, does not contributed to better seizure control than lesionectomy alone [44]. Extended lesionectomy is reserved for specialized cases in which concomitant focal cortical dysplasia is suspected [30].
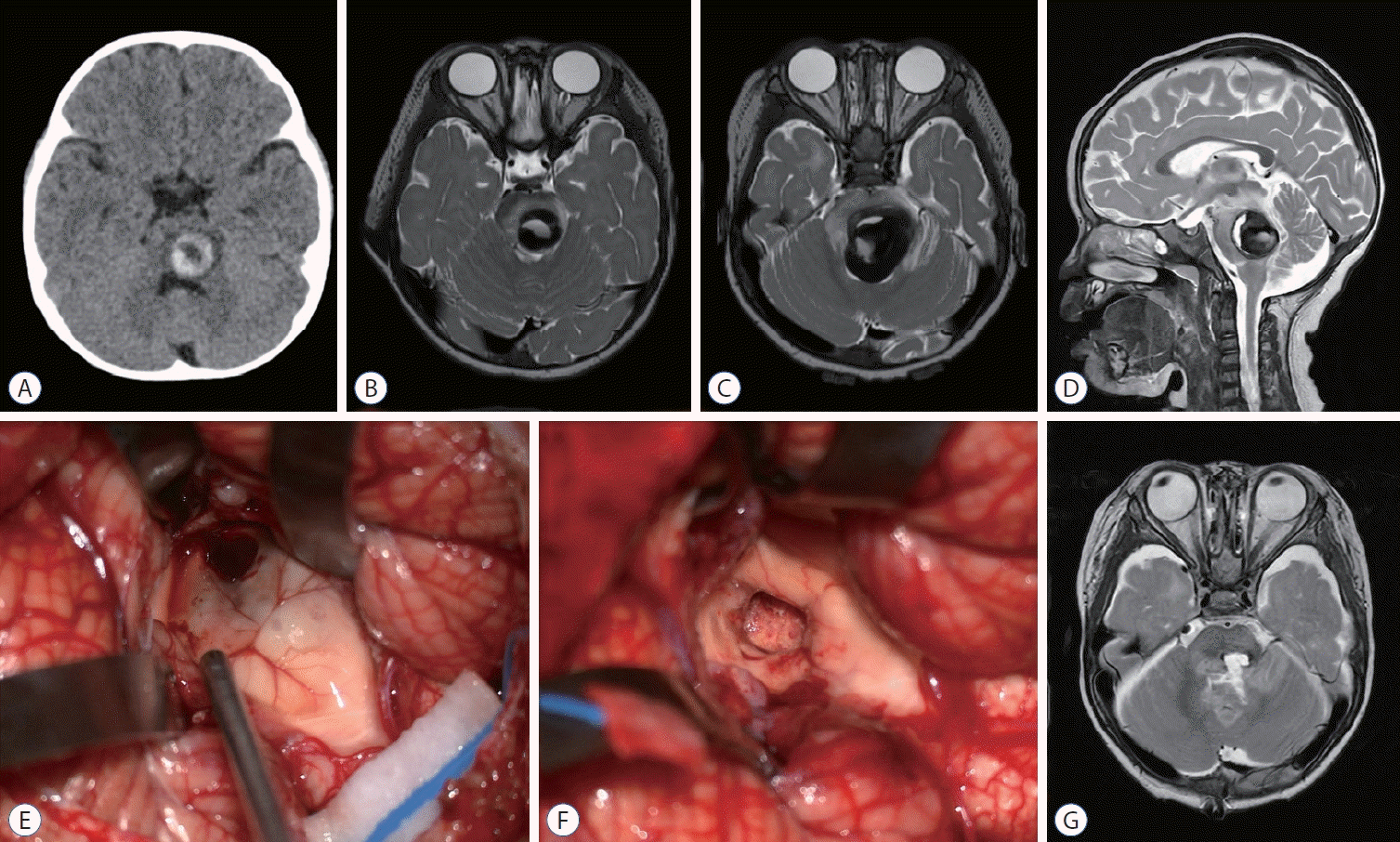
Brainstem CCM3 pose a particular challenge for neurosurgeons. They have a higher hemorrhage rate and are more likely to cause symptoms (Fig. 2). Stepwise deterioration can be observed in patients who experience repeated hemorrhages from a brainstem CCM. Brainstem CCMs can be approached when the lesion is exposed or come very close to the surface of the brainstem [38]. A two-point method was proposed that an ideal surgical approach should follow a line connecting the center of a CCM and the closest surface point [38]. Intraoperative monitoring is helpful in identifying safe entry zones, as the anatomy of the brainstem may be distorted if there is a large underlying mass [17]. Mapping of the fourth ventricle floor is valuable for identifying critical landmarks such as midline sulcus, facial colliculus, and hypoglossal and vagal trigones [34]. The hemorsiderin-stained gliotic rim should be preserved for brainstem CCMs, and caution should be exercised to preserve any associated DVA [17].
Stereotactic radiosurgery (SRS)
For patient with a deep-seated CCM, such as those in the brainstem, surgical risks and complication rates are high. In such cases, SRS, particularly gamma knife radiosurgery (GKRS), can be considered as an alternative option for preventing subsequent hemorrhages and neurological decline [26]. In a study involving adult patients, the AHR of CCMs dropped from 15.3% before treatment to 2.6% during the first 2 years from GKRS [25]. This reduction in AHR was observed across all locations, including the cerebrum and brainstem. The initial experience of GKRS for CCMs resulted in relatively high rate of complications (approximately 20%), but radiation-related complication have decreased with modifications in radiation dose and techniques [32]. Recent studies reported temporary complication rates around 15% and permanent deficits in 4–5% of patients [25]. A multi-center study on GKRS in pediatric patients demonstrated that AHR decreased from 7.2% before treatment to 3.1% post-treatment, with 14.5% developed adverse radiation effects; 55% of the patients had no or minimal symptoms [33].
Criticism persists regarding the gradual decrement of AHR observed in these SRS studies, as it may merely reflect the natural history of CCMs, considering the temporal clustering of hemorrhagic events [2]. CCMs are intrinsically dynamic lesions, with some growing and others shrinking over time. Hemorrhagic rates can vary greatly between individuals and over time in the same patient. Without a randomized clinical trial, controversy regarding the efficacy of SRS would continue [51].
Medical treatment
Identifying medications to enhance clinical outcome ranks among the top 10 research priorities for CCMs [5]. Medical treatment aims to prevent the de novo formation of CCMs, prevent CCM hemorrhage, and reduce the size of existing CCMs. However, no drug has been conclusively proven to achieve these goals in CCM treatment. Nonetheless, emerging evidence from epidemiological, preclinical, and clinical studies suggests that repurposing existing drugs or exploring novel target agents could benefit patients.
In a population-based study involving adult patients with CCM, the use of beta-blockers was associated with a reduced risk of new hemorrhage or persistent/progressive FND [56]. Conversely, female hormone therapy or oral contraceptive use was linked with a higher risk of CCM hemorrhages [57]. Preclinical studies have demonstrated that propranolol suppresses CCM formation in a zebrafish model with CCM2 gene disruption [29], and in a murine model with CCM3 gene-inducible knockout [35]. Recently published results from a randomized controlled phase 2 trial assessing the efficacy of propranolol for treating familial CCMs indicated a signal of efficacy, as the rate of new symptomatic hemorrhage or FNDs was lower in propranolol plus standard care group compared to the standard care alone (1.7 cases/100 person-years vs. 3.4 cases/100 person-years) [27].
The discovery of familial CCM-related genes, including CCM1, CCM2, and CCM3, has opened the door to the possibility of molecular target drugs for CCMs. The common biochemical derangement associated with the loss of CCM1, CCM2, and CCM3 is the activation of RhoA and increased Rho-associated coiled coil-forming kinase (ROCK) activity [41]. Fasudil, a specific ROCK inhibitor, has demonstrated the ability to suppress CCM development and prolong survival in a murine CCM model [46]. Statins, particularly atorvastatin, have shown efficacy in reducing lesion burden and hemorrhage in a murine CCM model by inhibiting ROCK activity [45]. Currently, a human randomized placebo-controlled trial is underway to evaluate the clinical impact of atorvastatin on CCMs [37].
CONCLUSION
CCM represents the most common cerebral vascular malformation requiring treatment in children and adolescents. However, the clinical courses of affected patients exhibit significant variability based on factors such as the patient’s age, lesion location, multiplicity, and family history. Additionally, the high and increasing proportion of asymptomatic patients further complicates the optimal management of CCM. Surgery remains the primary treatment modality for CCM. SRS presents an alternative option, particularly for deep-seated lesions that are not readily accessible; however, the efficacy of SRS requires further validation. Recent advancements in medical therapies offer promising avenues for non-invasive treatment and even prevention of CCMs in genetically predisposed children.
 XML Download
XML Download