

 PDF
PDF Citation
Citation Print
Print



Paramyotonia congenita (PMC) is a rare genetic disorder that is categorized within nondystrophic myopathy and is most commonly caused by variant.1 Clinically it is characterized by muscle stiffness exacerbated by cold exposure and paradoxical myotonia, and it usually shows autosomal dominance. Moreover, SCN4A-related channelopathy can manifest as PMC, sodium-channel myotonia, hyperkalemic or hypokalemic periodic paralysis, and (rarely) congenital myasthenic syndrome.1,2
From the genetic aspect, an autosomal dominantly inherited disease shows the same pathogenic variant in one of the parents. In rare cases a recurrent de novo pathogenic variant can be found, which may be explained by a germ-line mutation.3 Here we report the clinically classical manifestation of PMC in the presence of a pathogenic variant in SCN4A in two brothers, whose parents had wild-type SCN4A.
CASE
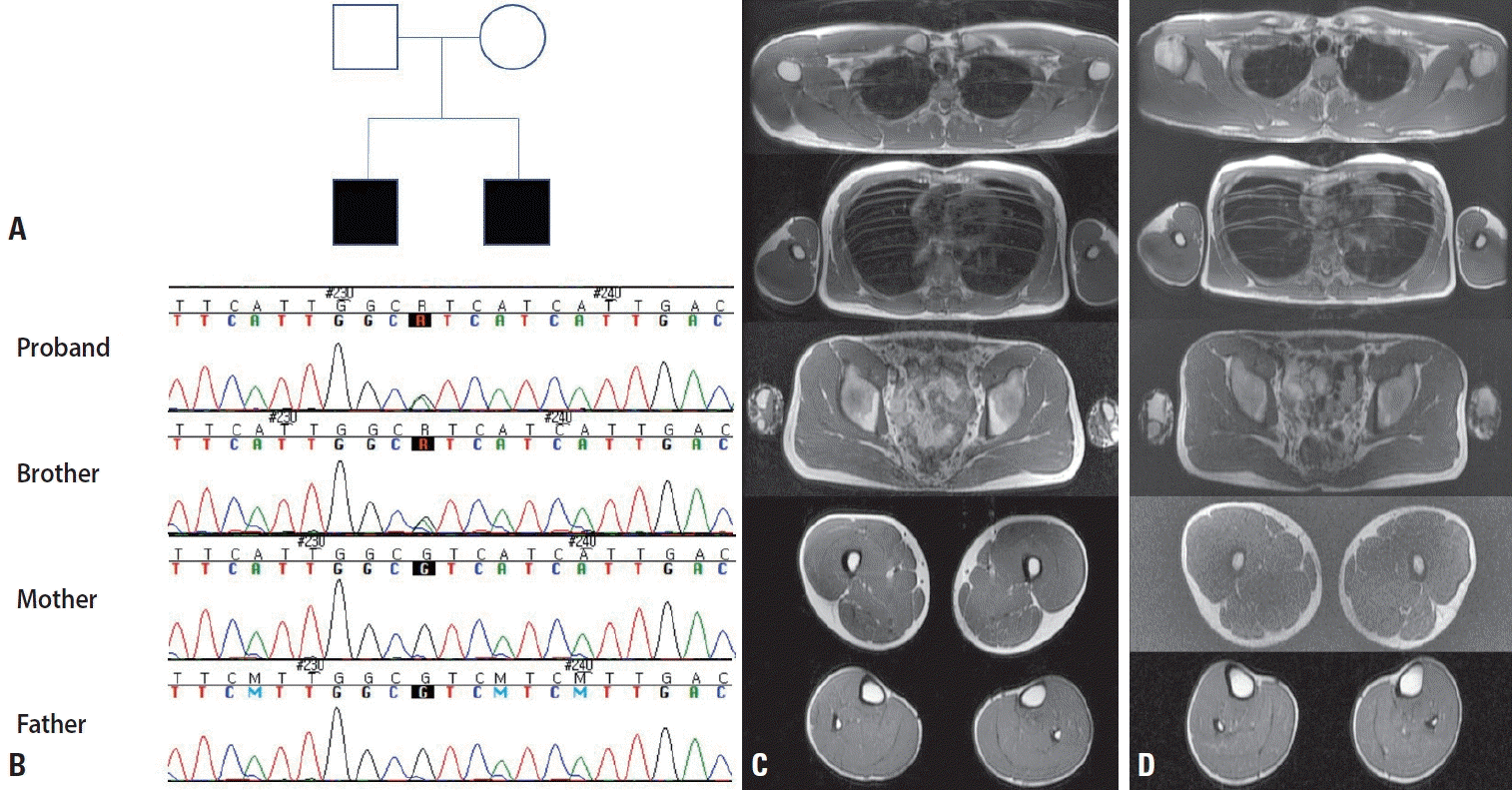
A 25-year-old male presented with intermittent muscle stiffness that was aggravated in cold weather since the age of 15 years. His younger brother had similar symptoms (Fig. 1A). The initial neurological examination showed no abnormalities in the cranial nerves or motor examinations, but the patient showed clinical myotonia in both hands that was aggravated by exercise. The deep tendon reflexes were normoactive with no abnormalities in the sensory symptoms. The routine laboratory findings were unremarkable, including for electrolytes, creatine kinase, and thyroid function. The nerve conduction study was unremarkable, but needle electromyography showed electrical myotonia in all tested muscles. Muscle magnetic resonance imaging (MRI) did not show significant radiological fatty change, but mild diffuse muscle hypertrophy was evident (Fig. 1C). The ice-pack provocation test significantly aggravated the clinical myotonia in the eyelids and hand. Targeted next-generation sequencing revealed the known pathogenic variant c.3877G>A, p.V1293I of SCN4A (Fig. 1B).
The 21-year-old brother of the proband also complained of muscle stiffness since the age of 17 years that was aggravated in cold weather. He had a clinical history of being unable to sing (especially in winter) due to difficulty in moving his mouth. The initial neurological examination was unremarkable, including of the cranial nerves and the motor and sensory systems. The routine laboratory findings encompassing electrolytes, thyroid function, and creatine kinase were within the normal ranges. An electrophysiological study produced normal nerve conduction findings, but electrical myotonia was observed in needle electromyography. Muscle MRI showed mild muscle hypertrophy, rather than muscle atrophy, as was present in the proband (Fig. 1D). He also showed paradoxical myotonia in his eyelids and hand that were clinically compatible with PMC. Sanger sequencing revealed the identical pathogenic variant carried by the proband (Fig. 1B). The parents of these two affected brothers were evaluated. The neurological examinations were unremarkable and neither of them exhibited clinical or electrical myotonia. To our surprise, blood-based Sanger sequencing did not detect the pathogenic variant that had been detected in their offspring (Fig. 1B). The final diagnosis was PMC with a pathogenic variant in the two brothers, with unaffected parents confirmed by genetic testing via blood sampling.
DISCUSSION
An SCN4A pathogenic variant can cause nondystrophic myotonia (NDM) and periodic paralysis, which makes it challenging in real-world presentations to segregate the genotype and phenotype correlation especially in the presence of SCN4A mutations.2,4 This gene encodes the alpha subunit of sodium channels that are abundant in muscles, and defects of this protein can lead to various types of ion-channel disease, including PMC, myotonia congenita, sodium-channel myotonia, hyperkalemic or hypokalemic periodic paralysis, and congenital myasthenic syndrome. Among SCN4A-related ion channelopathies, NDM is notorious for showing diverse phenotypes for the same mutation. The c.3877G>A variant present in our case was first described in 1995 by Koch et al.1 in three German families with autosomal dominant traits. All of those affected patients showed aggravation of muscle relaxation during cold exposure or paradoxical myotonia, which were clinically compatible with PMC. It is particularly interesting that a recent Korean myotonia congenita patient2 carrying the same pathogenic variant as our case showed abnormal relaxation and stiffness in the hand muscles that attenuated with repeated exercise; these manifestations were clinically compatible with myotonia congenita. These cases together reflect the broad clinical phenotype that can manifest in the presence of the same SCN4A variant even at the same site. From the genetic perspective, reported pathogenic variants of SCN4A are known to harbor missense mutations and usually show an autosomal dominant inheritance.5 SCN4A-related PMC can manifest clinically as a broad spectrum of clinical features ranging from myalgia and cramps without clinical myotonia to classical paradoxical myotonia and periodic paralysis.6
The primary pathomechanisms induced by SCN4A pathogenic mutations are mainly due to the rapid inactivation of sodium channels that cause myotonia, which can worsen with cold exposure. However, certain environmental aggravating factors such as exercise and cold exposure are known to influence these clinical phenotypes.
The presence of the same heterozygous pathogenic variant in our two brothers despite this variant being absent in both parents may be explained as a recurrent de novo case. However, a de novo case in two brothers is highly unlikely, and so attributing the condition to parental gonadal mosaicism may be more clinical possibility.3 Moreover, gonadal mosaicism has been reported in cases of different autosomal dominant or X-linked inherited genetic diseases.7-10
Autosomal dominant progressive external ophthalmoplegia has been reported in three siblings who harbored a pathogenic variant in ANT1, with no pathogenic variant found in either parent.7 A microsatellite analysis revealed a germ-line mosaicism in the mother. Autosomal dominant osteogenesis imperfecta (OI) was found in a symptomatic fetus in a family with a history of OI due to COLIA1 mutation, but with negative results in the parents in blood-based genetic testing.10 However, the genetic analysis revealed germ-line mosaicism in the sperm of the father. The main limitation of our case is that although we suspected the presence of gonadal mosaicism, we were unable to confirm this by performing a paternity test or germ-line testing of the sperm of the proband’s father, and hence caution is required when interpreting the present findings.
In de novo cases of channelopathies, especially when they are present in multiple siblings while the parents are unaffected in blood-based genetic testing, clinicians should consider the possibility of gonadal mosaicism, which might facilitate appropriate genetic counseling.
 XML Download
XML Download