

 PDF
PDF Citation
Citation Print
Print



Spinal muscular atrophy with lower limb predominance (SMA-LED) is characterized by congenital or early childhood onset, muscular weakness predominantly affecting the proximal lower limbs, and slow or absent progression.1 Two genes (DYNC1H1 and BICD2 genes) have been reported to be associated with SMA-LED.2 We report a patient presenting the clinical phenotype of SMA-LED concurrent with a germ-cell tumor who was carrying a pathogenic missense variant in DYNC1H1. This is the first study suggesting the association of a DYNC1H1 variant with the occurrence of a germ-cell tumor. Furthermore, this is the first case report of a patient carrying a DYNC1H1 variant who exhibited the typical clinical features of SMA-LED in Korea.
CASE
The male patient was born after 40 weeks of gestation. His family history was unremarkable (Fig. 1A), and the pregnancy and his delivery were uncomplicated. His early motor milestones were normal, and he could walk unassisted at 1 year old. However, an unstable gait developed at 3 years old and persisted thereafter. He was unable to run more than 50 m or climb stairs without assistance, and fell frequently. There was no progression of these symptoms.
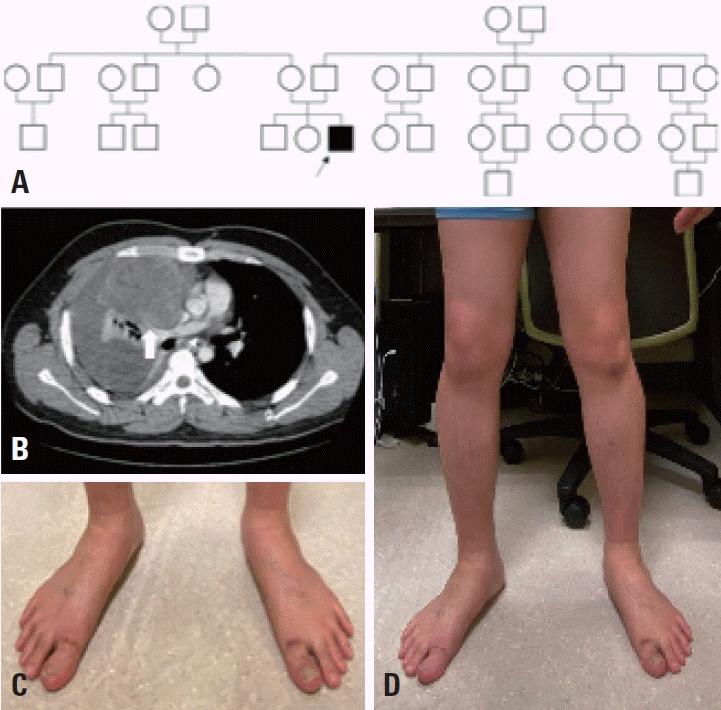
Fig. 1.
Pedigree and clinical features of the patient. (A) Pedigree of the three-generation family with dominant spinal muscular atrophy with lower extremity predominance. Filled symbol pointed by the arrow indicates the affected patient. There were no affected individuals other than our patient. (B) A heterogeneously enhanced mass of size 9.3×7.6×8.9 cm was found in the right mediastinum, and diagnosed as a mixed germ-cell tumor (arrow). (C) Foot deformity (talipes). (D) Distal wasting in lower limbs.
![]()
The patient visited our hospital when he was 25 years old due to chest pain. Chest computed tomography showed a heterogeneously enhanced mass in the right anterior mediastinum and right lung (Fig. 1B). Following cytomorphologic and immunohistochemical studies he was diagnosed with a mixed germ-cell tumor. He underwent resection for a mediastinal germ-cell tumor and received bleomycin, etoposide, and cisplatin chemotherapy for several months. He also visited our neuromuscular clinic because of his unstable gait. These symptoms were not aggravated after chemotherapy. He could walk unassisted but showed a waddling gait. A neurologic examination revealed decreased strength in both hip muscles of Medical Research Council grade 4+/5. Weakness of distal muscles including both calf and intrinsic hand muscles were less prominent. He could not rise from the floor without pushing with the arms, and had difficulty hopping. Vibration sensations at the malleoli were impaired, and the deep tendon reflex in both knees and ankles showed areflexia. There was also diffuse atrophy in the lower limbs and foot deformity (Fig. 1C, D). In contrast, his arms showed preserved muscle strength. He exhibited normal intellectual development. A clinical laboratory workup produced normal findings for the full blood count, creatine kinase (79 U/L), liver function, electrolytes, thyroid-stimulating hormone, and lactic acid levels. Brain magnetic resonance imaging also produced normal findings except for the presence of mild brain atrophy.
Nerve conduction studies revealed low sensory nerve action potentials bilaterally in the median, ulnar, peroneal, and posterior tibial nerves, which were probably due to the chemotherapy (especially cisplatin). Needle electromyography of the tibialis anterior, medial gastrocnemius, vastus lateralis, and tensor fasciae latae showed both denervation potentials and giant, long, polyphasic motor-unit action potentials, consistent with a chronic neurogenic process. A muscle biopsy performed when he was 3 years old had demonstrated severe atrophy of type-2 fibers, with marked increases in the amounts of fat and fibrous tissues, and sparse type-1 fibers.
We performed targeted next-generation sequencing of 160 genes for hereditary neuromuscular disorders (Supplementary Table 1) after obtaining informed consent from the patient. Unfortunately, genetic testing of the parents could not be carried out. The patient carried a heterozygous variant c.1793G>T in exon 8 of DYNC1H1, causing an arginine-to-leucine substitution (p.Arg598Leu), which was validated by Sanger sequencing, thus confirming the diagnosis of SMA-LED. In accordance with the American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines, the variant was classified as pathogenic. The same amino-acid change as a pathogenic variant was previously reported in a patient with Charcot-Marie-Tooth disease (CMT).3 The mutation located at the mutational hotspot and the variant were absent from a race-matched population. Multiple lines of computational evidence obtained from Polyphen-2 and SIFT support a deleterious effect on the gene product with high conservation. This variant may cause perturbations in tertiary structures with functional consequences due to differences between basic arginine and nonpolar hydrophobic leucine. The different substitution of arginine 598 to cysteine (p.Arg598Cys) reported in a patient with SMA-LED suggests that arginine 598 is important for protein function.3,4 Furthermore, MUpro server and I-Mutant2.0 predict that this DYNC1H1 mutation decreases the protein stability.5,6 Additionally, this variant was predicted in 2015 to be pathogenic.3 We concluded that this missense pathogenic variant affected the present case.
DISCUSSION
DYNC1H1 dysfunction can affect not only the development of the central or peripheral nervous system, but also the minus-end-directed transportation of cargo along microtubules. In particular, cytoplasmic dynein plays essential roles in the transport of numerous cell-cycle, apoptosis, and DNA-repair proteins from the cytoplasm to the nucleus.7 Here we have reported a patient presenting lower extremity predominant proximal weakness with early childhood onset, compatible with the clinical phenotype of SMA-LED. A heterozygous variant c.1793G>T in exon 8 of DYNC1H1, causing an arginine-to-leucine substitution (p.Arg598Leu), was identified as being causative. Unfortunately, genetic testing of the parents could not be carried out. However, given the absence of individuals within the family exhibiting gait disturbances, other neurologic abnormalities, or developmental decline, we consider it highly likely that the present case was a de novo occurrence.
DYNC1H1 has been reported to be associated with SMALED. A different substitution of arginine 598 was previously reported in a patient with SMA-LED, and there has been only one reported instance of the same amino-acid change as the pathogenic variant in a CMT patient.3 A single study from Korea highlighted a patient with SMA-LED who exhibited certain atypical clinical features, raising suspicions of hereditary spastic paraplegia and cognitive decline.8 Therefore, the present report is the first in Korea of this variant in a patient exhibiting the typical clinical profile of SMA-LED.
Cytoplasmic dynein regulates mitotic spindle localization and the connection of centromeric DNA to spindles during cell division.9 This indicates that DYNC1H1 dysfunction is associated with tumorigenesis. These observations can explain why altered expression of DYNC1H1 has been reported in various types of cancers, including of bone and the liver, gastrointestinal tract, and pancreas.9,10 We detected that the mutation was located in the stem domain, which is important in cargo binding and dynein heavy-chain dimerization. Also, the pathogenic variant is located close to the motor domain where deleterious variants are clustered, and lies across that domain in the folded structure. We therefore suggest that this variant was responsible for the tumorigenesis in this patient.
We suggest that this pathogenic variant of DYNC1H1 is correlated with both SMA-LED and germ-cell tumors. Clinicians need to consider DYNC1H1-related disorders as the causative genetic disease when a patient presents with both early childhood onset of muscle weakness and cancer.
 XML Download
XML Download