

 PDF
PDF Citation
Citation Print
Print



Introduction
Müllerian agenesis, also known as Mayer-Rokitansky-Küster-Hauser syndrome (MRKH), is a congenital disorder characterized by agenesis/aplasia of the Müllerian ducts during fetal development. MRKH syndrome is generally asymptomatic and is often diagnosed after puberty when the patient presents with primary amenorrhea, an inability to perform sexual intercourse, or dyspareunia. Women with MRKH syndrome typically exhibit the 46, XX karyotype. The external genitalia appear normal. However, the vagina is short, blind-ended, and may appear as dimples below the urethra. A uterine remnant may be present, with or without an endometrial cavity. Ovaries are typically normal in structure and function given their separate embryologic origins. Therefore, secondary sexual characteristics develop normally [1]. MRKH syndrome is a rare disorder with an estimated prevalence of 1:5,000 in women [2]. Despite its rarity, MRKH syndrome is the second most common cause of primary amenorrhea, following ovarian failure [3]. MRKH can be classified as type I (isolated) or type II (associated with malformations of extragenital organs involving the kidneys and skeleton) [2,4–6].
The initial evaluation of an adolescent with primary amenorrhea includes a physical examination, measurement of estradiol and follicle-stimulating hormone levels to assess ovarian function, chromosomal analysis to confirm a normal female karyotype, and imaging to assess the presence of the uterus [7]. Ultrasonography (USG) is the preferred initial diagnostic modality because it is simple and noninvasive [1]. Rudimentary Müllerian structures are found in 90% of patients with Müllerian agenesis on magnetic resonance imaging (MRI). Furthermore, MRI permits simultaneous evaluation of other associated anomalies, especially those of the urinary tract and skeletal system. Moreover, MRI can be used to assess the presence of endometrial activity within the Müllerian structures [8]. Laparoscopy is rarely indicated for diagnostic purposes and should be reserved for surgical interventions such as uterine remnant excision [1].
The management of Müllerian agenesis includes evaluation of associated congenital anomalies, interventions to improve vaginal length, and psychosocial counseling [7]. Müllerian agenesis negatively impacts sexual and psychological health. Women with this disorder may have lower self-esteem due to their inability to conceive as well as their inability to perform sexual intercourse [9,10].
The clinical presentation and extragenital organ malformations in MRKH syndrome differ among previous epidemiological studies from European countries and China [6,11–17]. Reports from Western countries and China have demonstrated heterogeneity in the phenotypic manifestations, including the types and rates of associated malformations, suggesting substantial ethnic differences. This study aimed to describe the clinical presentation, diagnosis, spectrum of associated congenital malformations, and management of Müllerian agenesis in a Thai population.
Materials and methods
This study was approved by the Institutional Review Board of the Faculty of Medicine at Songklanagarind Hospital (Institutional Review Board number: REC. 65-367-12-1). The database of all patients diagnosed with Müllerian agenesis between January 1, 2000 and October 31, 2022, was retrieved from the hospital information system of Songklanagarind Hospital, a tertiary referral center in southern Thailand. All medical records were reviewed and patients whose medical records could not be obtained were excluded from the analysis.
1. Identification of patients with Müllerian agenesis
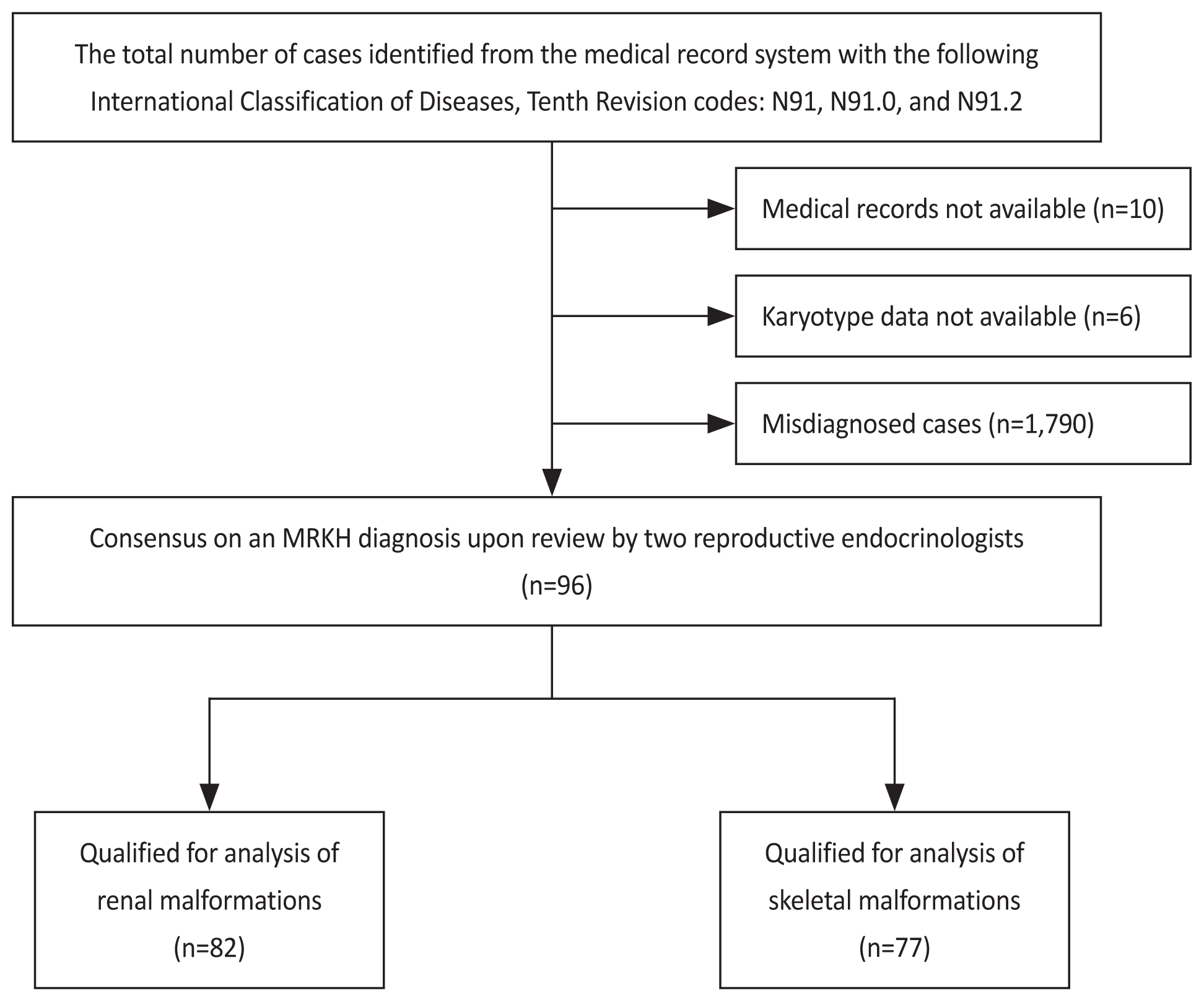
We identified women with Müllerian agenesis by reviewing the medical records of patients with the following International Classification of Diseases 10th Revision diagnosis codes: N91, amenorrhea; N91.0, primary amenorrhea; N91.2, amenorrhea, unspecified; Q51.0, agenesis, and aplasia of the uterus; Q51.5, agenesis, and aplasia of the cervix; Q51.8, other congenital malformations of the uterus and cervix; and Q52.0, congenital absence of the vagina.
The definitive diagnosis of Müllerian agenesis was retrospectively reviewed by consensus between two experienced reproductive endocrinologists based on the presence of the following features: primary amenorrhea with uterine agenesis/aplasia, normal profile of reproductive hormones or normal secondary sexual characteristics, and normal female karyotype (46, XX). Patients with no available karyotype data or those with the 46, XY karyotype were excluded from the analysis (Fig. 1).
Routinely performed diagnostic procedures included a thorough gynecological examination, pelvic USG, MRI, conventional karyotyping, and hormonal tests, including follicle-stimulating hormone and estradiol. Serum total testosterone levels were investigated solely in patients with hyperandrogenic symptoms. Associated extragenital anomalies involving the kidneys were evaluated using renal USG, intravenous pyelography, and MRI. Spinal abnormalities were identified using plain radiography or MRI. Patients with suspected cardiac or neuronal malformations were further examined by echocardiography, audiometry, or imaging-based tests. Organ malformations were classified according to the vagina cervix uterus adnex-associated malformation (VCUAM) classification.
Results
The medical records of 102 patients with MRKH syndrome who were diagnosed and treated during the study period were reviewed. Six patients were excluded due to missing karyotype results. The characteristics of the 96 patients are summarized in Table 1. The median patient age at diagnosis was 21 years. Only six patients (6.5%) were diagnosed before 15 years of age. The most common symptom was primary amenorrhea (88.5%), followed by difficulty or inability to engage in sexual intercourse (9.4%), and the presence of a pelvic mass (2.1%).
Pelvic examinations were not performed in 18 patients (18.8%) because they had never experienced sexual intercourse. More than half of the patients (52.0%) had undergone hormonal investigations, and all of these revealed normal estrogen levels (median of 72.6, interquartile range 51.7–220.9 pg/mL). All the remaining patients with no hormonal results had normal secondary sexual characteristics. The majority of patients were investigated for the presence of a uterus via transabdominal USG (83.3%), followed by transvaginal USG (16.7%), and both ultrasonography methods (5.2%). MRI was performed in 37.5% of the patients. Karyotype analysis was performed in all patients and revealed a normal karyotype of 46, XX, except for one patient whose karyotype test showed 46, XX, and 21p12+. This patient was a 16-year-old woman with an unremarkable family history. Further investigations revealed no extragenital malformations.
1. VCUAM classification
Clinical examination of the vagina was performed in 78 patients, revealing a stage 5b (complete atresia) vagina in 21.8% of the patients, whereas 78.2% of the patients displayed a stage 4 (hypoplasia) vagina. None of the patients had a cervix. Imaging results revealed 85 patients (88.5%) with stage 4b (bilateral rudimentary or aplastic), two patients (2.1%) with stage 4a (unilateral rudimentary or aplastic), and nine patients with stage 3 (hypoplastic uterus) uterine anomalies. None of the images showed functional endometrial activity within the Müllerian structures. None of the patients showed abnormal adnexal masses on imaging.
Regarding malformations associated with MRKH syndrome (Table 2), 76 of 96 patients (79.0%) were completely evaluated using both urinary tract and skeletal imaging. Furthermore, 61 of the 76 patients (80.3%) had no extragenital malformations and were diagnosed with MRKH type I (typical form), whereas 15 of the 76 patients (19.7%) were diagnosed with MRKH type II (atypical form). Only one patient had a combination of malformations (renal and skeletal). Skeletal malformations were the most frequent extragenital anomalies (15/77, 19.5%), with scoliosis being the most common skeletal abnormality, followed by renal alterations (7/82, 8.5%). A neurological abnormality was found in one patient, diagnosed with congenital anomalies of the craniovertebral junction. One patient exhibited abnormally located ovaries.
2. Treatment
Half of the patients included in this study received no treatment because they had not engaged in sexual intercourse. Among the cohort, 41.7% had no difficulty performing sexual intercourse; thus, self-dilatation therapy or coital dilatation was advised. Only eight patients (8.3%) underwent surgical reconstruction of the vagina owing to the failure of primary dilatation therapy or their preference for surgery.
Discussion
This study included 96 women diagnosed with MRKH syndrome according to the VCUAM classification. The clinical presentation of MRKH syndrome is complex and highly heterogeneous, with varying rates and degrees of vaginal atresia and extragenital malformations. In this study, the rate of extragenital malformations was 19.7%, with skeletal abnormalities being the most common.
In our cohort, the median age at the time of diagnosis was 21 years, and this is consistent with previous studies that have reported the average age at the time of diagnosis to range from 17.2–26.2 years [4,11,12,14–17]. The late age at the time of diagnosis may be explained by the fact that women with MRKH syndrome usually present with primary amenorrhea without abdominal pain. Moreover, they displayed normal pubertal development, including growth spurt and breast development. Therefore, the absence of menstruation may not be perceived as a problem by these women. Patients with MRKH syndrome generally present with primary amenorrhea, inability to engage in sexual intercourse, and dyspareunia. Occasionally, they may also present with cyclical pelvic pain due to functional endometrial activity within the remnants of the Müllerian structures [1]. Primary amenorrhea was the most common symptom in our cohort, followed by the inability to engage in sexual intercourse and dyspareunia. Interestingly, two patients in our cohort (aged 39 and 51 years) presented with palpable abdominal masses. They had well-developed secondary sexual characteristics, but had never commenced menstruation. Pelvic examination revealed a blind-ended vaginal pouch in both the individuals. Chromosomal analysis revealed the 46, XX karyotype in both patients. The provisional diagnosis was solid ovarian tumor in both cases, and the patient underwent surgical excision. However, exploratory laparotomy revealed rudimentary uteri with fibroids; the histological diagnoses were uterine leiomyoma and adenomyosis arising from the rudimentary uterus in both cases. The presence of leiomyoma and adenomyosis in MRKH syndrome is rare, and only a few cases have been reported in the literature [18,19]. However, the exact pathogenesis of leiomyomas or adenomyoses in the rudimentary uterus of patients with MRKH syndrome remains unclear.
Evaluation of adolescents with primary amenorrhea includes hormonal measurements to assess ovarian function, chromosomal analysis, and imaging to assess the presence of a uterus [7]. The estradiol levels in our study were within the normal range, indicating normal ovarian function due to their separate embryological development. However, their anatomical position has been noted to be more cranial, probably owing to the lack of fallopian tube development [4]. We found that 1% of the patients had ovarian abnormalities (abnormally located ovaries), which is concordant with the findings of Wang et al. [20] who reported a higher incidence of abnormally located ovaries in patients with MRKH syndrome. Previously reported ovarian anomalies include hypoplastic or aplastic ovaries [12,13].
The choice of imaging modality employed, including USG and MRI, differed between patients in our study. Most patients underwent transabdominal USG because they had not previously engaged in sexual intercourse or had complete vaginal atresia. If the USG images were inconclusive, or if the clinician suspected an obstructed rudimentary uterus, MRI was performed. Uterine remnants were reported in 6.2% and 41.7% of our patients, as detected by transabdominal USG and MRI, respectively, which was lower than that reported in previous studies indicating uterine remnants in 48–95% of patients [11,21,22]. Moreover, none of the patients exhibited functional endometrial activity.
Chromosomal analysis was necessary to exclude a diagnosis of androgen insensitivity syndrome (AIS). AIS generally presents with primary amenorrhea, a shortened vagina, and an absent cervix. Moreover, patients with AIS have normal breast development owing to the aromatization of testosterone to estrogen. We found that normal 46, XX karyotypes were present in 99% of cases in this cohort, with an abnormal karyotype being present in one case (46, XX, and 21p12+), and this rate was similar to that reported in previous studies (0.3–1.4%) [4,13,15,16]. The patient with the abnormal karyotype had presented with amenorrhea and an unremarkable family history. Further investigation revealed no extragenital malformations. Previous studies have revealed aberrations in different chromosomal regions, including 1q21.1, 16p11.2, 17q12, and 22q11.21 microduplications and deletions in patients with MRKH [23]. To the best of our knowledge, 21p chromosomal variants have never been reported to be associated with MRKH syndrome. Overall, these findings suggest the presence of chromosomal aberrations in patients with MRKH syndrome.
Regarding extragenital malformations, this study showed that only 19.7% of the patients had atypical MRKH syndrome (type II). Previous large epidemiological studies on MRKH syndrome were predominantly based on data from European countries [6,11–15], with two studies from China [16,17]. Type II MRKH syndrome accounted for 27.8–55.4% of the participants from Europe, which was higher than that in the Chinese population (7.2–28.1%) (Table 3). These findings suggest that there are substantial differences across ethnicities [17]. The rate of extragenital malformations was also lower in our study (19.7%) compared to that in the European population [6,11–15]. Moreover, skeletal malformations were the most common abnormalities in our study (19.5%), in contrast to most previous studies that reported renal abnormalities as the most common extragenital malformations [4,11–16]. This difference in the incidence and types of extragenital malformations could be due to selection bias (single-center case series), incomplete data on malformations, or ethnicity. To the best of our knowledge, the congenital anomalies of the craniovertebral junction reported here in one patient have not been previously described in association with MRKH syndrome.
Patients with MRKH syndrome require a functional vagina to improve their quality of life, particularly their sexual function. Complete vaginal atresia was observed in 21.8% of our cohort; however, this rate was lower than that reported in previous studies, where complete vaginal atresia was present in 100% of the patients with MRKH syndrome [12,16]. Phenotypic manifestations also appear to vary in terms of the severity of vaginal atresia. The treatment recommended for patients with MRKH syndrome is psychological counseling and support upon diagnosis as well as creating a functional vagina to improve sexual function. Most patients in our cohort were advised regarding self-dilatation therapy or coital dilatation according to the recommendations of the American College of Obstetricians and Gynecologists committee [7]. Vaginal elongation by dilatation is the appropriate first-line approach in most patients because it is safer and generally achieves satisfactory results with a low risk of complications. However, surgery should be reserved for patients for whom primary dilatation therapy has failed or who primarily prefer surgery.
Our study has several limitations, including its retrospective design and the varying imaging modalities used among the patients. Moreover, 20.8% of patients remained unclassified because of a lack of urinary tract and/or skeletal imaging. Therefore, the number of patients with atypical MRKH syndrome might have been underestimated.
In conclusion, our study confirmed the complexity and heterogeneity of the MRKH phenotypic manifestations, including the degree of vaginal atresia and the types and rates of associated malformations.
 XML Download
XML Download