

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Breast cancer is the most common malignancy among women worldwide [1]. Due to changes in lifestyle and an increasing proportion of an aging population, the incidence of breast cancer is expected to rise in Asian countries. The National Comprehensive Cancer Network commends biennial breast cancer screening through mammography for women over 40 years old. However, mammography showed limited diagnostic screening value in women with dense breasts; improved screening methods are needed as early diagnosis could be missed by mammography only.
Breast cancer is known to be influenced by genetic and environmental factors, and susceptibility genes including BRCA1/2, PALB2, and ATM have been discovered. Still, the majority of genetic contributors to breast cancer risk remains unknown. Recent studies have focused on translating genomics, proteomics, and precision medicine to validate further risk factors and potential screening biomarkers that could be applied easily to the general population [23].
Proteomic profiling of human plasma is a novel technique for cancer biomarker discovery [4]. The protein profile in distinct tissues shows substantial heterogeneity that does not fully reflect the complexity of the whole proteome, which acts as a limitation to clinical application [5]. In contrast, plasma proteome contains secreted proteins originating from multiple organs, providing a basis for a more comprehensive biomarker discovery. Blood component analysis is the most widespread diagnostic procedure in clinical practice, and biomarkers detectable in blood plasma could be easily integrated into national screening programs [6].
This study aimed to compare the plasma proteome of 10 breast cancer patients and 10 healthy individuals by mass spectrometry (MS) to identify potential differences in protein expression profiles.
METHODS
Study population
This study was conducted as a pilot study using bioresources from the Human Bioresource Center of Seoul National University Bundang Hospital (No. DT-2020-012-01). Serum samples from 10 healthy individuals and 10 breast cancer patients were obtained for this study by random selection. Baseline characteristics of the study participants were provided by the Human Bioresource Center; any information related to patient identification was not available.
Plasma sample preparation
A multiple affinity removal column (MARS-14, 4.6 × 100 mm, Agilent) was used to remove human plasma abundant proteins. A 40-µL aliquot of plasma diluted with 160-µL Agilent buffer A was filtered through a 0.22-µm cellulose acetate filter to remove particles. The diluted plasma sample was injected into a MARS-14 depletion column on a binary high-performance liquid chromatography (LC) system (20A Prominence, Shimadzu). The unbound fraction was collected into a collection tube and completely dried using a speed-vac concentrator (Thermo Fisher Scientific). The dried sample was resuspended in 100 µL of S-Trap lysis buffer and sonicated for 10 minutes. Depleted plasma was followed by reduction with 10-mM dithiothreitol at 56 ℃ for 30 minutes and alkylation with 20-mM iodoacetamide at room temperature for 30 minutes in the dark. Samples were then prepared with S-Trap spin column (S-Trap mini, ProtiFi) based tryptic digestion according to the manufacturer’s instructions. A microspectrophotometer (Allsheng) was used for tryptic peptide quantification, and samples were frozen at –75 ℃ until use.
Liquid chromatography-mass spectrometry/mass spectrometry analysis
Plasma peptides were separated using a Dionex UltiMate 3000 Rapid Separation Liquid Chromatography (RSLC) nano system (Thermo Fisher Scientific). The tryptic peptides were separated on an Acclaim PepMap RSLC C18 column (150 mm× 150 µm, inner diameter [i.d.] of 2 µm, 100 Å; Thermo Fisher Scientific) equipped with a C18 PepMap trap column (20 mm × 100 µm, i.d. of 5 µm, 100 Å; Thermo Fisher Scientific) over 60 minutes (1 µL/min) using a 5%–40% acetonitrile gradient in 0.1% formic acid and 5% dimethyl sulfoxide at 50 ℃. The LC system was coupled to an Orbitrap Exploris 480 mass spectrometer with an EASY-SPRAY source (Thermo Fisher Scientific). For data-independent acquisition (DIA) experiments, full MS scan resolutions were set to 60,000 and the automatic gain control (AGC) target was 300% with an injection time (IT) of 25 ms. The m/z range was set to 300–1,400. The MS/MS spectra scan resolution was set to 15,000 and the AGC target was 10,00% with an IT of 22 ms. Forty-four windows of 24 Da were used with an overlap of 1 Da. The normalized collision energy of 27 was used for higher-energy collisional dissociation. The MS/MS scan range was set to 300–1,800.
Data-independent acquisition analysis and data processing
The DIA MS data were processed using DIA-NN (ver. 1.8) with library-free mode [7]. Spectra were searched with default settings except match-between-runs activated. Identification results were filtered at a false discovery rate of 1% at the precursor level. Protein quantities were obtained using the MaxLFQ algorithm [8].
Gene set enrichment analysis
Gene set enrichment analysis was performed using ConsensusPathDB [9]. We restricted our analysis to gene ontology biological processes (GOBPs) and pathway databases including Kyoto Encyclopedia of Genes and Genomes, Reactome, and WikiPathways [101112]. Only the GOBPs and pathways with the number of molecules involved ≥10 were retained.
Differential expression analysis
We defined differentially expressed proteins (DEPs) using an integrative statistical method previously reported [13]. We calculated test statistics using the Student t-test, Wilcoxon rank-sum test, and a log2-median-ratio in each comparison for protein expression. We then estimated empirical distributions of the test statistics and log2-median-ratios for the null hypothesis by randomly permutating all samples 1,000 times. Using the estimated empirical distributions, for each protein, we computed adjusted P-values for the observed test statistics and log2-median-ratio. Finally, we defined DEPs as the proteins that had the Student t-test P-values <0.05 and absolute log2-median-ratios greater than the mean of the 10th and 90th percentile of the empirical distribution for log2-median-ratios in each comparison. Only the proteins that were expressed in more than half of the total samples in both testing groups were used.
RESULTS
Clinicopathological characteristics
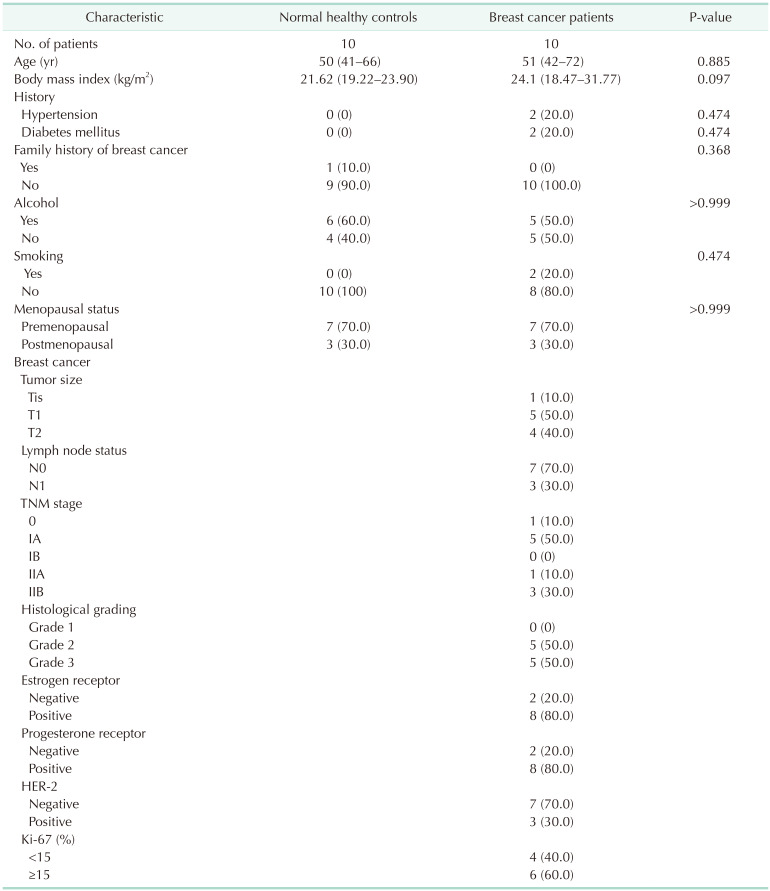
The clinicopathological characteristics of the study participants are summarized in Table 1. All included participants were women. There were no statistically significant differences in age, body mass index, past medical history, family history of breast cancer, social history, and menopausal status between the breast cancer patients and healthy controls included in the analysis. Pathological information was available only for breast cancer patients. It included tumor size, lymph node status, TNM stage, histological grade, estrogen receptor status, progesterone receptor status, human epidermal growth factor receptor-2 (HER2) status, and Ki-67 index.
Evaluation of plasma mass spectrometry data identification results
We performed quantitative plasma proteome profiling of 10 breast cancer patients and 10 healthy controls in the cohort for breast cancer diagnostic biomarker discovery. The DIA-NN search resulted in the identification of 6,702 peptides and 891 proteins across all samples, and 882 and 805 proteins were identified from normal and breast cancer groups, respectively. Half of the identified proteins were annotated plasma proteins in the Human Protein Atlas database (https://www.proteinatlas.org) (Fig. 1A). The identified plasma proteome was estimated to cover approximately 7 orders of dynamic ranges: 4.4E11 pg/L of ceruloplasmin to 9.2E4 pg/L of hippocalcin-like protein 1 (Fig. 1B).
Functional enrichment of the breast cancer plasma proteome
Principal component analysis showed that the identified plasma proteome clearly separated breast cancer patients from healthy controls (Fig. 1C). All identified proteins were used for gene set enrichment analysis. We found breast cancer-related GOBPs including blood coagulation, cytoskeleton organization, cytokine-mediated signaling pathway, and regulation of the cell cycle. In addition, pathway analysis revealed that membrane trafficking, focal adhesion, glycolysis and gluconeogenesis, vascular endothelial growth factor A (VEGF-A)-VEGF receptor 2 signaling pathway, vesicle-mediated transport, regulation of insulin-like growth factor (IGF) transport and uptake by IGF-binding proteins, and tight junction were significantly enriched by the breast cancer plasma proteome (Fig. 1D). Next, we performed differential expression analysis to identify potential diagnostic biomarkers for breast cancer. We identified 30 upregulated and 100 downregulated proteins in the plasma of breast cancer patients compared to healthy controls (Fig. 1E, Supplementary Table 1).
Potential biomarker selection
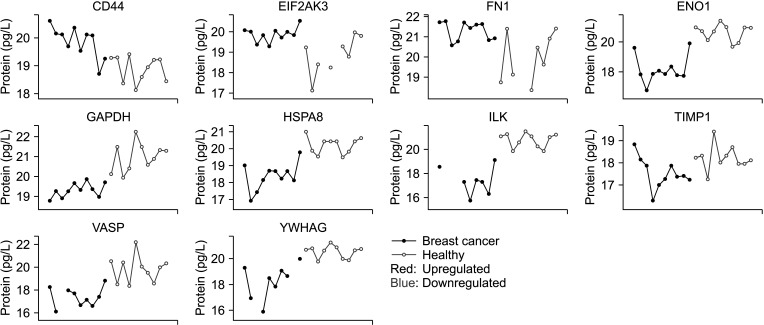
Among the 130 proteins differentially expressed between the plasma of breast cancer patients and healthy controls, 10 proteins reported to be differentially regulated in breast cancer by previous studies were selected. The 10 proteins were chosen as representative examples of proteins with known involvement in GOBP or signaling pathways and biological functions associated with breast cancer reported by prior studies. Cluster of differentiation (CD) 44, eukaryotic translation initiation factor 2-α kinase 3 (EIF2AK3), and fibronectin 1 (FN1) were upregulated in breast cancer patients. Seven proteins that were downregulated were also selected: glyceraldehyde-3-phosphate dehydrogenase (GAPDH), α-enolase (ENO1), heat shock protein member 8 (HSPA8), integrin-linked kinase (ILK), tissue inhibitor of metalloproteinases-1 (TIMP1), vasodilator-stimulated phosphoprotein (VASP), and 4-3-3 protein gamma (YWHAG). The expression pattern of the selected proteins was consistently up- or downregulated across all cancer samples, with a few cases showing overlapping expression levels with those of healthy control samples (Fig. 2).
DISCUSSION
Plasma biomarkers play an essential role in the screening, diagnosis, and follow-up of malignancies. Plasma is easy to obtain through standardized procedures at a relatively low cost and effort, and plasma proteome is known to reflect diverse tissue proteome subsets [6]. Proteomics allows for comparative profiling of DEPs between diseased and control samples or at various stages of progressive disease [14]. Therefore, biomarkers identified through plasma proteomic profiling could be applied for diverse purposes including early diagnosis and therapeutic response monitoring.
For breast cancer, several tumor biomarkers have been proposed, including cancer antigen 15-3, CEA, HER-2, and tissue polypeptide-specific antigen [1516]. However, the clinical utility of these markers has been limited by their low sensitivity and specificity. Recent studies have focused on the analysis of breast cancer plasma proteome for identification of DEPs [1417]. Discovery of novel biomarkers through advanced proteomics techniques is promising, especially for screening purposes, as the false negative rate for mammography reaches 10%–30% worldwide.
We analyzed the plasma proteome of 10 breast cancer patients and 10 healthy controls by MS to identify differences in protein expression profile. We found 891 proteins across all samples, of which 805 were expressed in the breast cancer group and 882 proteins in the control group. Differential expression analysis revealed that 30 proteins were upregulated and 100 were downregulated in breast cancer patients’ plasma compared to those of healthy controls. Analysis of the plasma proteome presents technical challenges, as human plasma contains a diverse range of protein concentrations. Although numerous proteins related to general biological and tumor-related processes were identified, only 10 proteins were chosen as potential biomarkers. All selected proteins had been reported in preexisting literature to be differentially expressed in breast cancer patients, which suggests that they may be directly or indirectly involved in the disease’s development.
CD44, EIF2AK3, and FN1 showed upregulation in breast cancer patients. CD44 is an adhesion molecule that plays a role in tethering cells to the extracellular matrix. Aberrant expression of CD44 has been noted to play a role in the metastasis of breast tumors, as well as in cancer stem cells [18]. EIF2AK3 is a transmembrane protein that acts as a stress sensor in the endoplasmic reticulum. It has been recognized as a crucial mediator in estrogen receptor stress-induced autophagy, which prevents cancer cell apoptosis [19]. EIF2AK3-dependent signaling triggers various processes in the metastatic cascade, including angiogenesis, cell migration, and colonization at secondary organ sites. Fibronectins participate in cell adhesion and migration processes, and previous studies have shown that FN1 is involved in the development of thyroid cancer, renal cell carcinoma, and nasopharyngeal cancer [20]. Zhang et al. [21] found that FN1 was upregulated in breast cancer tissues at both messenger RNA (mRNA) and protein levels, with higher FN1 mRNA expression correlated with poor prognosis.
Seven proteins that were downregulated in breast cancer patients were also selected as potential plasma biomarkers. GAPDH is a glycolytic enzyme with non-glycolytic functions including regulation of cell death, autophagy, and DNA repair. The role of GAPDH in cancer cells seems to be complex, and both overexpression and suppression of the protein have been suggested as possible mechanisms of tumor progression [22]. For the other 6 biomarker candidates that were suppressed in breast cancer patients in the current study, these results were contradictory to previous literature. Although these proteins are well-known oncogenes in cancer, we did not rule them out as potential biomarkers for breast cancer based solely on their putative contradictory patterns between blood expressions and oncogenic functions at the tissue level. Instead, given that the blood concentrations of these proteins can also be influenced by various factors besides direct secretion from breast cancer cells, we included them in our list of putative breast cancer biomarkers based on their observed blood expression patterns in our data and their reported functional significance in breast cancer.
ENO1 plays an essential role in cell growth, hypoxia tolerance, and tumorigenesis, and its glycolytic function sustains tumor proliferation and inhibits apoptosis. Overexpression of ENO1 in breast cancer tissues has been linked with poor prognosis [23]. The 70 kDa heat shock protein (HSP70) is associated with tumor proliferation and metastasis. HSPA8, one of the important members of HSP70, has recently been proposed as a new biomarker for triple-negative breast cancer [24]. The role of ILK has been widely studied in breast cancer, and silencing of ILK leads to apoptosis and decreased cell invasion [25]. On the other hand, ILK overexpression promotes tumor growth and metastasis [26]. TIMP1 has a tumor-promoting function via growth stimulation and inhibition of apoptosis. High plasma levels of TIMP1 have also been associated with poor response to hormonal therapy and chemotherapy [27]. VASP promotes cell migration in many malignant diseases including gastric cancer and cervical cancer; the expression level of VASP shows a positive correlation with advanced tumor stage in lung cancer; and in hepatocellular carcinoma, VASP promotes tumor invasion and metastasis. In breast cancer, VASP is a target gene of Wnt/β-catenin pathway, and its activation leads to cell proliferation and migration [28]. YWHAG, also known as 14-3-3γ, is an oncogenic target protein related to pseudopodia, which accounts for cell motility in breast cancer [29]. Upregulation of YWHAG has been linked to increased tumor proliferation and metastasis in pancreatic cancer and lung adenocarcinoma [30]. As the expression levels of these proteins were downregulated in the current analysis, further studies on their role in breast cancer are warranted.
This study has certain limitations. First, the study was limited by its retrospective nature. Second, the relatively small sample size limited the conclusions of the study. As this was a pilot study to identify potential proteome biomarkers, a future validation study with a larger population of both breast cancer patients and healthy controls is necessary to further investigate the predictive power of these novel biomarker candidates. We are currently planning to validate the results of this study with a larger cohort of breast cancer patients from the same institution, including analysis for differential protein expression according to breast cancer subtype.
In conclusion, we found that the plasma proteome profile differed significantly between breast cancer patients and healthy controls; based on these results, we identified 10 potential plasma biomarkers for breast cancer screening. The incidence of breast cancer is rising and several contributing genes have been identified; however, there is still an unfulfilled demand for clinically applicable biomarkers. The current study provides the cornerstone for the identification of biomarkers easily detected through routine serological examinations.

 XML Download
XML Download