

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Antimicrobials stand out as one of the most remarkable medical discoveries of the 20th century. Their application in the healthcare sector has averted millions of premature deaths from infectious diseases (1). Since the advent of the first antibiotic in 1910, there has been an estimated average increase of 23 years in human life expectancy (2). However, its widespread and indiscriminate use in the past decades resulted in the emergence of highly drug-resistant pathogenic microorganisms.
Antimicrobial resistance (AMR) is defined as the ability of microorganisms to withstand the effects of agents and drugs designed to kill them (3). AMR has the potential to greatly impair the clinical sector, life expectancy, and food production, making it one of the most serious global health concerns in modern times. If left unaddressed, the World Health Organization (WHO) estimates that AMR could cause 10 million deaths per year by 2050 (4) and a cumulative global economic loss of more than $100 trillion (5). The economic costs of AMR are detailed in the review by Pulingam et al. (6).
The COVID-19 pandemic has exacerbated AMR due to the increased misuse and overuse of antibiotics in healthcare settings (7). This surge in antimicrobial use has compromised the efficacy of many frontline antibiotics. Furthermore, unchecked secondary bacterial and fungal infections during the pandemic have accelerated AMR development. This poses a severe threat to individual treatment outcomes and global public health, with projections indicating that future AMR-related deaths could exceed those caused by COVID-19 (8). Additionally, the uncontrolled environmental release of antimicrobials and biocides during the pandemic is likely to further accelerate the global growth of AMR (9).
Although AMR is a blanket term used to describe resistance discovered in microorganisms such as viruses, parasites, fungi, and bacteria, this review will primarily focus on bacterial antibiotic resistance. AMR can be classified into two types: intrinsic resistance and acquired resistance. Some bacterial species are naturally resistant to antibiotics because of certain inherited properties, or they simply do not possess the target for the antibiotic compound. In Gram-negative bacteria, for example, lipopolysaccharides in their cell walls provide an effective barrier against the penetration of certain antibiotics, thus limiting the uptake of drugs (10). In contrast, acquired resistance pertains to initially susceptible bacteria that gained a trait or developed a mechanism that enables it to evade the effects of the antibiotics. This type of resistance may emerge through spontaneous mutations or via the acquisition of resistance genes. Generally, bacteria acquire these genes through transformation (assimilation of resistant genes from the environment), transduction (gene acquisition from bacteriophages), and conjugation (transfer of genes from resistant bacterial strains via sex pili) (11). The number of effective antimicrobial agents that are available has been severely constrained due to the emergence of resistance in pathogenic bacteria to several antimicrobial drugs. Hence, a standardized categorization of those with multiple resistance was established by the Centers for Disease Control and Prevention (CDC) and European Centre for Disease Prevention and Control (ECDC) relevant to the field of health and epidemiology. Multidrug resistance (MDR) refers to the non-susceptibility of a microorganism to one or more antimicrobial agents from at least three different classes. On the other hand, extensive extensively drug-resistance (XDR) refers to the non-susceptibility to at least one agent in most classes of antimicrobials (12).
The impact of AMR is heightened by the limited progress in the search for novel antimicrobial medications (13). An antibiotic drug is considered novel based on the following criteria: (a) belongs to a novel chemical class and interacts with new molecular targets/binding sites, (b) works through new mechanisms, and/or (c) biochemically modified in such a way that a previously resistant pathogen is resensitized (14, 15). Currently, the number of active programs searching for novel antibiotics is scarce (16). Furthermore, it is widely observed that resistance to an antibiotic often emerges shortly after a novel drug has been introduced for clinical use. This was evident with penicillin, as bacterial strains capable of producing penicillinase—an extracellular enzyme degrading penicillin—were reported shortly after its introduction as a therapeutic agent for bacterial infections. It is important to note that penicillin and its derivatives (cephalosporins and carbapenems) are still the predominant class of antibiotics used to treat human and animal bacterial infections to this day (17).
Bacterial resistance poses a significant challenge due to their alarmingly rapid adaptation. Studies show they can acquire resistance to new antibiotics in just a few years (18). This rapid evolution is driven by the widespread use of antimicrobials in various sectors, including healthcare and agriculture (19), highlighting the need for proactive measures to combat this threat. To mitigate this risk, a multifaceted approach must be taken. Stewardship programs that advocate for judicious antibiotic use are essential to minimize selection pressure on bacterial populations (20). Additionally, surveillance programs are crucial to monitor emerging resistance trends and inform the development and deployment of future antimicrobials (21). While numerous alternative therapies like phage therapy and microbiome modulation offer hope in combating the growing threat of antibiotic-resistant bacteria (22), in particular molecular approaches like nanomaterials and clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 hold particular promise due to their unique advantages over traditional antibiotics. These approaches offer two benefits: enhanced efficacy and reduced potential for resistance development. Nanomaterials improve drug delivery and cellular uptake, potentially revitalizing the effectiveness of existing antibiotics (23). CRISPR-Cas9, with its precise targeting of essential bacterial genes, disrupts critical functions and hinders the development of resistance (24). This review provides an update on the recent progress and understanding of the genetic and molecular mechanisms of antibiotic resistance. Emerging molecular strategies in addressing antibiotic resistance and challenges and advancements in treating MDR pathogens are also discussed.
ANTIBIOTIC RESISTANCE MECHANISMS
Antibiotics are effective in treating bacterial infections. These agents can alter metabolic and physiological levels, causing modifications in cell wall synthesis, protein synthesis inhibition, metabolic pathway inhibition, and DNA interference (25). However, factors such as (a) misuse by physicians and patients, (b) inappropriate diagnosis and prescription, and (c) overuse contribute to antibiotic resistance in healthcare (26). In addition, incomplete antibiotic use can lead to the development of resistant strains, posing a serious public health concern and emphasizing the need for cautious use (27).
Bacterial cells exposed to antimicrobial agents can be either resistant or persister. Non-resistant cells are killed, leaving only resistant ones. When regrown, all cells become resistant. Non-persister cells are killed, leaving only persister cells, which are susceptible to the antimicrobial agent when regrown. One widely accepted reason for such evasion against antimicrobial agents is due to dormancy. In a review of Wood et al. (28), a small fraction of bacterial cells in a population undergoes a dormant stage, a metabolically inactive state, found to be linked with the stress response mediated by the toxin-antitoxin system, as one mechanism. Resistant cells, often caused by genetic mutations or resistance genes, are less susceptible to specific antibiotics due to their mechanisms. Genetic mutations alter antibiotic targets, while horizontal gene transfer spreads resistance genes (29). Efflux pumps expel antibiotics, while enzymatic inactivation renders drugs inert (30). Resistant cells pose a persistent challenge, necessitating alternative antibiotics or combination therapies to address infections caused by these resistant strains. Persister cells, on the other hand, temporarily exhibit antibiotic tolerance, often characterized by metabolic dormancy or slowed growth (31). They do not have genetic changes but enter a reversible physiological state that shields them from the immediate effects of antibiotics (32). Resistant cells require new antimicrobial agents, targeted therapies, and strict infection control measures, while persister cells require extended treatment regimens (33). The complex nature of microbial responses to antibiotics underscores the need for more innovative strategies to manage infections caused by rapidly evolving antibiotic resistant pathogens.
In the last few years, several antibiotic resistance mechanisms (including enzymatic degradation, target site alteration, efflux pumps, reduced permeability, biofilm formation, and genetic mutations) were found to be common across different reviews and studies (34, 35, 36, 37, 38). One of the many strategies used by bacteria is the synthesis of enzymes, specifically β-lactamases, which can break down or alter antibiotics such as cephalosporins and penicillin. The effectiveness of these medications is neutralized by this enzymatic change, which also plays a major role in the emergence of antibiotic-resistant bacteria (39). Additionally, bacteria demonstrate their adaptability by changing the locations of their targets, which lessens their vulnerability to the effects of antibiotics. Target site modifications have led to the discovery of multiple genetic variants that result in antibiotic resistance in various classes of antibiotics. Fluoroquinolones, for instance, induce mutations in the genes encoding bacterial DNA gyrase and topoisomerase, leading to a reduction in their binding affinity. These compounds target these enzymes (40).
Bacteria develop a high level resistance to antimicrobial agents by reducing their entrance (41). Efflux pumps, acting as molecular exporters, actively remove substances and drugs from bacterial cells, contributing significantly to antibiotic resistance. They can give bacteria resistance to a wide range of medications, which helps bacterial infections develop MDR (42). Recent studies into the structural and functional characteristics of efflux pumps have shown a variety of pump families with particular substrates. Bacteria have the potential to possess multiple efflux pump systems capable of eliminating specific classes of antibiotics. Studies also demonstrate how bacteria can dynamically modify their pump activity in reaction to stimuli from their surroundings or the presence of antibiotics (30, 34). Another way that bacteria resist the effects of antibiotics is by having less permeability in their cell membranes. Furthermore, resistance is increased by the development of biofilms, which are complex colonies of microorganisms encased in an extracellular matrix for protection (43). By limiting the diffusion of antimicrobial drugs and consequently blocking their intended targets within bacterial cells, the matrix functions as a physical barrier, shielding bacterial cells from immunological and environmental stresses as well as inhibiting antibiotic penetration (44, 45).
Genetic mutations can play a role in antibiotic resistance because they can result in changes to vital cellular components that drugs target due to spontaneous changes in bacterial DNA (46). These mutations change the nucleotide sequence during DNA replication, especially in genes that encode antibiotic target sites (47). Through structural modifications, these mutations can impart resistance to target proteins like fluoroquinolones (48). Furthermore, horizontal transfer of resistance genes plays a critical role in the rapid spread of antibiotic resistance within bacterial communities and the propagation of resistance features across species. This occurs when bacteria conjugate, transform, or transduce resistance features from other living things (29). Additionally, mutations can impact the uptake and efflux of antibiotics by altering the permeability of cell membranes or strengthening efflux pumps, both of which can lower the concentration of antibiotics inside cells (49). Because of this dynamic interplay, the environment of resistance is continuously changing, requiring in-depth research to understand mutation patterns, trace evolutionary pathways, and create new defenses against antibiotic- resistant bacteria.
Types of Resistance to Antibiotic Agents
Understanding the systems responsible for resistance is vital in the development of antimicrobial drugs that avoid the development of resistance and expand available therapeutic options. The majority of pathogenic bacteria are capable of becoming resistant to some antimicrobial treatments. Antibiotic resistance can be intrinsic, acquired, or adaptive (34, 50, 51).
Natural resistance can either be intrinsic or induced. Intrinsic resistance, which may be attributed to evolution (50), refers to the resistance that a bacterium exhibits as a result of its natural characteristics (52). Intrinsic resistance is universally shared within a species, independent of antibiotic exposure. Some bacteria may naturally produce enzymes that break down or prevent drugs from binding to target structures causing “insensitivity” (27). Other common mechanisms include reduced outer membrane permeability and efflux pumps, while induced resistance involves multidrug-efflux pumps (34).
Most pathogenic microorganisms can develop resistance to certain drugs, with main mechanisms involving limitation of drug uptake, modification of drug targets, inactivation, and active efflux. Understanding these mechanisms could lead to improved treatment options and the development of antimicrobial drugs that can withstand resistance attempts by microorganisms (27).
Bacteria can acquire resistance to antimicrobial agents through genetic mutations, assimilating DNA from resistant bacteria, altering protein production, and transferring resistant DNA through horizontal gene transfer and direct contact (50). Helicobacter pylori poses a treatment challenge for gastrointestinal diseases due to its ability to develop antibiotic resistance via mutations in its 23S rRNA gene. These mutations hinder antibiotic binding, allowing H. pylori survival and potentially compromising treatment success. In staphylococcal species, these mutations reduce susceptibility to linezolid, which has led to resistance in Staphylococcus aureus and Streptococcus pneumoniae but remains effective in over 98% of Staphylococcus species. Ali et al. (27) presented how mutations may lead to antibiotics being unable to bind to their target.
Microbial species that are resistant to antibiotics need to pass on their resistance genes to their offspring (vertical transmission) and be able to transmit genes horizontally—that is, through transformation, transduction, and conjugation—to preserve their resistance. Transformation involves transferring DNA fragments from a dead bacterium to a recipient bacterium, while transduction refers to the transfer between donor and recipient bacterium. Conjugation is another process that involves the transfer of genetic material between bacterial cells through physical contact. This process may include multiple resistance genes on a single plasmid and is facilitated by mobile genetic elements (52).
Adaptive resistance is a conditional, unstable, and transient microbial resistance caused by a specific signal or environmental condition, which is not vertically inherited and can save costs for genetic modification. One mechanism for adaptive resistance is the formation of biofilms, Biofilms are associated with persistent chronic infections and are now a main target for developing antimicrobial agents (51). Antimicrobial agents selectively target crucial microbial functions and exhibit varying effects. These agents are classified into distinct groups based on their mechanisms of action such as interference with cell wall synthesis, membrane depolarization, inhibition of protein synthesis, suppression of nucleic acid synthesis, and inhibition of bacterial metabolic pathways (26, 34).
The bacterial cell envelope, which consists of the plasma membrane and cell wall, provides structural integrity and protection from internal pressure. The cell wall, an elastic macromolecule, is responsible for maintaining cell shape and protecting bacteria from lysis, making it an ideal target for antibiotic therapy. Antibiotics like β-lactams and glycopeptides, inhibit cell wall synthesis through two mechanisms. β-lactams target penicillin-binding proteins (PBPs), which are bacterial proteins that bind to penicillin and other antibiotics of the β-lactam class (26, 53). Meanwhile, glycopeptides kill Gram-positive bacteria such as methicillin-resistant Staphylococcus aureus (MRSA) and Enterococcus species by disrupting peptidoglycan layers (26, 53). Resistance to β-lactams may arise in two ways: by changing the structures of PBPs to make them less attractive to antibiotics, or by generating β-lactamase, an enzyme that breaks down the β-lactam ring and renders antibiotics inactive (54). The decline in antibiotic susceptibility observed in S. aureus and the emergence of glycopeptide- resistant enterococci, attributed to mutations in transpeptidase enzymes, pose considerable health risks. In 2002, the isolation of vancomycin-resistant S. aureus (VRSA) strains occurred following the horizontal transmission of a resistant gene from Enterococcus faecalis to MRSA in vivo. Moreover, multiple proteins encoded on mobile transposons are the cause of inactivating the vancomycin mechanism (26, 55).
The peptidoglycan outside the cytoplasmic membrane acts as a permeability barrier. Polymyxin antibiotic molecules attract negatively charged bacteria, making the cell membrane more permeable, leading to osmotic imbalance and cell death (55). Additionally, antibiotics, such as macrolides, aminoglycosides, and tetracycline, can selectively inhibit protein synthesis due to structural differences between bacterial and eukaryotic ribosomes. Chloramphenicol can bind to the 50S subunit of the bacterial ribosome, preventing new polypeptide addition (34, 56). Resistance to aminoglycosides is carried out through ribosomal mutations, ribosomal methyltransferase enzymes, and aminoglycoside-modifying enzymes. Ribosome mutations can alter the 16S subunit of the ribosome, potentially leading to lethal effects. Ribosomal methyltransferases in Actinomycetes protect ribosomes from aminoglycosides by methylating the 16S subunit. Aminoglycoside-modifying enzymes are chemical modifications that result in aminoglycoside resistance, and their function is still uncertain but can be transmitted horizontally between bacteria through plasmids, integrons, or transposons (53, 57). Efflux pumps utilize active transport mechanisms to eliminate aminoglycosides from bacterial cells, albeit with limited effectiveness attributed to the polycationic structures of aminoglycosides (52, 53, 54). AcrAD, a multidrug transporter in gram-negative bacteria, works against aminoglycoside antibiotics (26).
Furthermore, three possible enzymatic modifications can lead to erythromycin resistance: (1) ereA or ereB genes in erythromycin, (2) mgt gene causing glycosylation in macrolides, and (3) mphA, mphB, and mphC genes causing macrolide phosphorylation (34, 57). In Escherichia coli, a sequence mutation alters the II and V domains, leading to erythromycin resistance. In S. aureus, methylation of A2058 in domain V can happen through two mechanisms: either by the action of the adenine N-methyltransferase enzyme or by a mutation that changes A2058 to guanine. These alterations lead to reduced antibiotic affinity (26). Macrolide efflux transporters can be divided into proton motive force pumps and ATP pumps, or by their structure to the single and complex integral membrane (57).
Fluoroquinolones, which are broad-spectrum antibiotics, cause mutations in DNA topoisomerase genes and inhibit DNA gyrase in Gram-negative bacteria, and topoisomerase IV in Gram-positive bacteria, leading to double-stranded DNA breakdown and cell death (34, 55). Bacterial resistance to fluoroquinolones is developed through two main mechanisms: enzyme alteration and drug-access alteration. Enzyme alteration involves DNA gyrase mutations, such as GyrA or GyrB subunits, which can cluster in the Quinolone Resistance Region and reduce drug affinity. Drug-access alteration involves MDR efflux pumps, which excrete drugs outside the cell before reaching the target site, reducing fluoroquinolone activity. The outer membranes of Gram-negative bacteria also provide more resistance due to reduced protein porins, lowering drug diffusion rate (26, 52, 53, 54).
Reduced folate co-factors are essential for various cellular components in prokaryotic and eukaryotic cells. Tetrahydrofolate is required in biosynthetic processes and degradative reactions. Trimethoprim and sulfonamides target the folate biosynthetic pathway, inhibiting para-aminobenzoic acid use in bacterial folate synthesis. These antibiotics are used to treat urinary tract infections (UTI), but resistance to these inhibitors has been detected (52). Trimethoprim and sulfonamides resistance are common in S. aureus and Streptococcus pneumoniae, resulting from amino acid substitutions in the dhfr gene and duplications in the folP gene, respectively, causing chromosomal alteration in the dhfr gene and tertiary structure of the enzyme (52). Meanwhile, rifampin resistance is due to a mutation in the rpoB gene, affecting the β-subunit in the RNA polymerase. It is common in bacteria like Mycobacterium tuberculosis, where it is the only active antibiotic used. Therefore, it is recommended to use it in combination with other antibiotics to prevent resistance (26, 56).
Antimicrobial-resistant microorganisms are more widespread due to misuse, incorrect dosing, and a lack of novel antibiotics. These pathogens can impair cancer chemotherapy, common ailments, surgical procedures, and organ transplants. The absence of pertinent data and the creation of new tools and technologies have a big impact on early diagnosis and treatment. In order to protect antibiotics for future generations, collective and proactive efforts at several social levels are required.
ANTIBIOTIC RESISTANCE IN THE CLINICAL FIELD
Antibiotic resistance has become particularly pronounced in clinical settings, as it poses a significant threat to global population health and can lead to reduced treatment options, resulting in higher costs for healthcare systems (58). The broader concept which is AMR is alarming as well as it results in close to 1.27 million deaths annually and it is crucial to note that this pattern is consistently increasing on a global scale. The six primary pathogens responsible for fatalities linked to resistance include E. coli, S. aureus, Klebsiella pneumoniae, S. pneumoniae, Acinetobacter baumannii, and Pseudomonas aeruginosa (5).
In the Annual Epidemiological Report in European Antimicrobial Resistance Surveillance Network (EARS-Net) for the period 2017 to 2021, there were 22 countries that submitted the report with the highest frequency for E. coli (accounting for 39.4% of all reported cases), succeeded by S. aureus (22.1%), K. pneumoniae (11.9%), E. faecalis (8.8%), Enterococcus faecium (6.2%), P. aeruginosa (6.1%), Acinetobacter spp. (3.0%), and S. pneumoniae (2.5%). The current order differs from that of 2020, with E. faecium and Acinetobacter spp. now holding one rank higher. The documentation of cases involving pathogens displaying AMR aligns with shifts in healthcare and community dynamics brought about by the COVID-19 pandemic. These changes are likely to have impacted activities related to infection prevention and control targeting these specific pathogens (59). As for the latest surveillance report of 2022, it was observed that in comparison to the previous year (2021), the aggregate number of reported isolates exhibited an increase from 366,794 to 392,602. Notably, E. coli was the most frequently reported bacterial species in 2022, constituting 39.2% of cases, followed by S. aureus (22.1%), K. pneumoniae (12.3%), E. faecalis (8.2%), P. aeruginosa (6.1%), E. faecium (5.9%), S. pneumoniae (3.7%), and Acinetobacter spp. (2.5%) (59).
Examining data specific to Southeast Asia, information from Bangladesh, Nepal, and Sri Lanka highlights a notable resistance prevalence to β-lactams, primarily attributed to the presence of pathogens producing extended-spectrum beta-lactamase (ESBL) (60, 61, 62). In the year 2021, A. baumannii demonstrated a resistance rate of 87.5% to carbapenems. Over the period from 2016 to 2021, there was a progressive increase in the rates of MRSA, rising from 28.4% to 42.6%. In the context of Indonesia, aligned with the UN Sustainable Development Goals (SDG) indicator, the proportion of bloodstream infections induced by ESBL-producing E. coli accounted for 57.7% of the total E. coli cases (63). Among children in Myanmar, elevated levels of carbapenem resistance were observed for E. coli (48%), K. pneumoniae (42%), and Acinetobacter spp. (59%) (64). In 2016, a study estimated that 43% of deaths (19,122 out of 45,209) in Thailand are associated individuals with infections that are hospital-acquired and were attributed to multidrug-resistant bacteria, which indicates a surplus of mortality linked to resistant pathogens (65).
In China, a similar investigation was undertaken to monitor AMR within its major hospitals. The quantity of strains collected in the year 2022 exhibited an increase compared to the statistics recorded in 2021. The predominant bacterial strains, namely E. coli, K. pneumoniae, S. aureus, P. aeruginosa, and A. baumannii, largely maintained their prevalence. The rate at which methicillin-resistant strains were detected experienced a continuous decrease. Regarding clinical Enterobacterales isolates, the resistance to carbapenems generally remained below 13%, with the exception of Klebsiella spp., where resistance ranged from 20.4% to 21.9%. The majority of clinical Enterobacterales isolates demonstrated notable susceptibility to tigecycline, colistin, and polymyxin B, with resistance rates ranging from 0.1% to 12.6%. Over the past four years, there has been a consistent decline in the detection rates of meropenem-resistant P. aeruginosa and meropenem-resistant A. baumannii (66).
The escalating global health threat of bacterial AMR necessitates urgent action. Thus, Murray et al. (5) gave the following recommendations. (a) Prioritize building laboratory infrastructure to address the significant and universal burden of AMR. (b) Improve management of individual patients and enhance the quality of data in local and global AMR surveillance. (c) Strengthen national AMR plans of action to combat AMR effectively on a broader scale. (d) Expand AMR research to evaluate indirect effects, including impacts on perioperative prophylaxis, prophylaxis in transplant recipients, transmission dynamics, and prevalence of specific variants through genotypic epidemiology. (e) Identify and implement strategies to reduce the burden of bacterial AMR, considering both general and context-specific settings, and tailoring interventions to available resources and leading pathogen-drug combinations. (f) Recognize the urgent priority of addressing bacterial AMR globally, with a focus on sub-Saharan Africa and South Asia, while acknowledging its importance in all regions. In the future, a more comprehensive and programmatic approach is essential to enhance the effectiveness of interventions against AMR. This approach, as highlighted by Sihombing et al. (67), emphasizes placing people and their needs at the core of the AMR response.
CHALLENGES AND ADVANCEMENTS IN TREATING CRITICAL MDR BACTERIA
When a bacterial strain acquires resistance to more than one type of antibiotic, it is called MDR bacterium (68). These strains are immune to various structures of antibiotics and to distinct mechanisms of action (68). The MDR arises from mutations, selection, and/or acquired resistance through horizontal gene transfer (68, 69). It has been an exigent emergency in public health as infections caused by MDR bacteria have killed millions worldwide (5, 21). Such strains of MDR bacteria, that are virtually immune to any medical interventions, are called ‘superbugs’ which are proven to be lethal and urgent concern (70). A number of critical superbugs are included in this chapter namely, (a) carbapenem-resistant Acinetobacter, (b) methillin-resistant S. aureus, (c) ESBL-producing Enterobacteriaceae, (d) MDR-P. aeruginosa, and (e) MDR-M. tuberculosis. In addition, challenges in treatment and advances in new classes of antibiotics, nanotechnology, phage therapy, and vaccines are tailored to provide present knowledge as well as future directions.
Carbapenem-resistant Acinetobacter baumannii
The WHO regarded carbapenem-resistant A. baumannii as one of the most critical priorities in developing new classes of antibiotics and treatments due to failure in antibacterial response of the current ones (21). A. baumannii is a Gram-negative coccobacillus regarded as a nosocomial pathogen (71). During the COVID-19 crisis, increased incidences of co-infections were documented in the intensive care units of Qom, Iran (72). One of the fitness advantages of A. baumannii is its natural resistance to most mechanisms of action of antibiotics. Its carbapenem resistance is attributed to acquiring carbapenemases. A novel type of siderophore cephalosporin named cefiderocol was designed to specifically target many species of Gram-negative bacteria (Enterobacteriaceae, especially P. aeruginosa and A. baumannii) by compromising cell wall biosynthesis while evading the β-lactam enzymes (73).
In a phase III clinical trial across 16 countries, cefiderocol demonstrated comparable efficacy to the best available therapy in treating nosocomial pneumonia, UTI, and bloodstream infections caused by MDR Gram-negative strains. It is recommended as an alternative medication, particularly for Acinetobacter spp. infections (74). Another relevant finding regarding those with nosocomial pneumonia is the option of utilizing cefiderocol aside from meropenem (75). However, both reports of Basetti et al. (74) and Wunderink et al. (75) administered a high dosage of the drug with 2 milligrams every 8 hours. In addition, the study of Portsmouth and colleagues provided the basis for the application of the drug against UTI during the phase II clinical trial. Cefiderocol, at a similar dosage, was found to be non-inferior against imipenem-cilastatin (76). Susceptibility to cefiderocol in A. baumannii may decrease due to a single amino acid substitution in penicillin-binding protein 3 (PBP3), as evidenced by one isolate post-treatment, resulting in a fourfold increase in the minimum inhibitory concentration (MIC) (77). Analyzing and comparing changes in the sequence and target protein of novel drug classes can offer valuable insights to enhance their efficacy and durability.
Colistin, a polymyxin antibiotic, has been found ineffective in both monotherapy and combination therapy with carbapenem against Gram-negative bacteria (78, 79, 80). Due to its limited efficacy and associated adverse effects such as diarrhea and renal impairment, its use should be avoided, especially in critically ill patients (78, 81). In response to the decrease in effective antibacterial strategies over time, there has been a shift from broad-spectrum to pathogen-focused drug development. Addressing carbapenem-resistant A. baumannii and Acinetobacter calcoaceticus, Watkins et al. (82) developed a structure-based drug targeting three predominant classes of β-lactamases (Class A, C, and D). The novel sulbactam-durlobactam (SUL-DUR), acting as a β-lactam and β-lactamase inhibitor, has received urgent approval from the United States Food and Drug Administration (FDA) this year. SUL-DUR has met the non-inferiority criterion and exhibited tolerable adverse effects and lower nephrotoxicity compared to colistin and the imipenem-cilastatin combination (82, 83). This promising new treatment option is now available in the United States for severe cases of MDR-A. baumannii (83).
The use of nanoparticles (NPs) is one of the avenues to combat the looming resistance against antibiotics. The conjugation of imipenem with silver nanoparticles (AgNPs) (10-40 nanometers) signified effectiveness during in vitro studies of clinical isolates in Iran, where 76% were initially resistant to imipenem alone (84). AgNPs alone were proven to mediate bacterial apoptosis and obstruct DNA synthesis as revealed by Chen et al. (85). Most of the reported applications of NPs are in vitro with promising low cytotoxicity levels. However, there are hypotheses and studies regarding the co-emergence of resistance against the wide use of AgNPs which must be considered for appropriate use. McNeily and others (86), demonstrated the increased tolerance upon prolonged exposure observed in A. baumannii. The aforementioned case demands further testing to ensure the appropriate use of NPs as well as verifying its translational application to animal and clinical systems. Recently, novel integration of gold NPs (AuNPs) and DNA aptamer for the delivery of the antimicrobial peptide showed promising reduction of A. baumannii proliferation in mice (87). Another strategy explored by Al-Kadmy et al. (88) is by interfering with the capsule and biofilm formation with the use of copper oxide NPs. They found a decrease in efflux pump synthesis upon exposure but still recommended for further evaluation especially regarding its clinical safety (89). Meanwhile, nanoengineered vaccine delivery of outer membrane vesicles with the use of AuNPs provided a proof- of-concept regarding its reliability and potential against pneumonia and sepsis (90). However, succeeding tests on mice with immune deficiency and other relevant human diseases must be cleared first prior to human trials (89). Developing vaccines dedicated to high-risk individuals can save more lives and simultaneously reduce reliance on antibiotics.
Methicillin-resistant Staphylococcus aureus (MRSA)
MRSA is originally a nosocomial pathogen which is prevalent now in the community which is automatically categorized as MDR (12). Its resistance to β-lactams is due to the gain of the mecA gene, which is responsible for the synthesis of penicillin- binding protein 2a (PBP2a) (90). In the review by Ambade et al. (91), targeting PBP2a with five- and six-membered heterocyclic compounds shows robust potential, yet integrating this knowledge while considering human microbiome and safety remains challenging. Linezolid, an oxazolidinone introduced in 2000, inhibits bacterial protein synthesis by binding to the 23S ribosomal subunit (92). Levonadifloxacin, a benzoquinolizine fluoroquinolone, has demonstrated superior cure rates against skin infections compared to linezolid and is now approved in India (92, 93). Ceftobiprole, a broad-spectrum new-generation cephalosporin, completed a phase III trial, proving non-inferiority to daptomycin (94). Although it has a relatively lower rate of adverse effects, gastrointestinal discomfort and nausea are common (94). Pharmacokinetic studies and clinical trials have shown its suitability for both Asian and non-Asian populations, as well as for pediatric patients (95, 96, 97). Contezolid, approved in China, outperforms linezolid in treating skin infections (98, 99).
Many bacterial infections may be treated using phage therapy; however, because lytic phages have a limited host range, most bacterial pathogens must be treated empirically with cocktails of phage strains that differ in their host ranges and virulence traits (100). Whittard and others established that four (4) among the 85 phages showed the best wide-range host potential, bacterial growth reduction, and biofilm inhibition against the globally distributed MRSA sequence types. Two phages (EW 41 and EW 71) are promising candidates to be studied coming from the genus Silviavirus and Kayvirus, respectively. One important consideration in developing phage therapy against MRSA is the possibility of horizontal gene transfer of prophages that may induce virulent toxins (100). Nonetheless, the option of phage utilization still lacks wide and authorized application.
There is still no vaccine available against hospital-acquired or community-acquired MRSA. However, one notable progress in this area is targeting cytotoxic alpha-hemolysin (Hla) delivered through epitope-based nanoparticles (HlaH35L) (101). Both the in vivo study in mice and human serological tests elicited neutralizing activities. However, the EpNP mode of vaccination proposed is still not the best candidate over the current alum-adjuvant vaccine (101). This continues to represent a groundbreaking approach in formulating a vaccine against MRSA that is set to enter clinical trials. It is recommended to evaluate the EpNP vaccine’s effect on immune cells and to incorporate other neutralizing epitopes.
Extended spectrum beta-lactamase (ESBL)-producing Enterobacteriaceae
Another critically urgent superbug considered are found in the family of Enterobacteriaceae with a special priority with the species of K. pneumoniae, Enterobacter spp., and E. coli (102). These species often exhibit extended-spectrum β-lactamase or carbapenem resistance, contributing to high mortality rates in hospital intensive care units (ICU) (102). Clinical trials in their final stages frequently employ combination therapies, where one drug eradicates bacteria while the other targets specific resistance mechanisms to enhance effectiveness. In a phase III clinical trial involving 268 participants with complicated intra-abdominal infection (cIAI), the use of ceftolozane/tazobactam plus metronidazole yielded a superior cure rate difference of 2.1% (95% confidence interval) compared to the meropenem group (103). This treatment regimen demonstrated safety, with no reported adverse effects, in contrast to the three cases observed in the meropenem group. Most importantly, this serves as another option in treating ESBL-positive Enterobacterales especially for K. pneumoniae and E. coli (103). The subgroup analysis by Patterson et al. (104) supported the better clinical outcome of ceftolozane/ tazobactam against the ESBL-positive and/or carbapenemase-producing Enterobacterales. The most common type present is the Cefotaxime-Munich (CTX-M-15) ESBL that also represents the global occurrence in the species of K. pneumoniae and E. coli (104, 105). The findings of Mikamo and others (106) in 100 Japanese cIAI patients coincide with the effective administration of ceftolozane/tazobactam with a clinical response rate of 92.0% with a baseline pathogen profile including both E. coli and K. pneumoniae. Hence, the most susceptible inhibitory mechanism must be identified or narrowed down first to have a higher chance of efficacy while ensuring the longevity of a designed drug or treatment in addressing the highly resistant Enterobacterales. In addition, a fourth generation with a broad-spectrum drug eravacycline, now FDA approved, has comparative efficacy as meropenem against MDR- and XDR-Enterobacterales (73, 107). Under clinical trials, eravacycline is non-inferior to meropenem with a better safety profile and low adverse effects (107). Meanwhile, there are no adequate and conclusive reports on the use of NPs in treating MDR-Enterobacterales. In one published in vitro study in a different susceptibility profile of K. pneumoniae (a clinical XDR strain), α-lipoic acid-capped AgNPs associated with imipenem synergistic effects in reducing the cell viability and biofilm formation (108). The study utilized the augmentation of the α-lipoic acid which minimized the required concentration of AgNPs without diminishing its effectiveness (108). As observed in many in vitro studies, the antimicrobial activity of metal oxides and metallic NPs stems from their accumulation in the bacterial cell wall, disrupting the organism’s physiological processes (109). The next crucial steps in advancing the field of nano-antibiotics include conducting pharmacokinetic and safety evaluations in animal models (109).
MDR-Pseudomonas aeruginosa
P. aeruginosa, a Gram-negative bacterium, is also included in the family of Enterobacterales (110). It is an opportunistic pathogen, highly implicated in the healthcare setting for its MDR profile, that complicates patients with existing conditions such as pneumonia, cystic fibrosis, skin wounds or damages, and UTI (110). MDR-P. aeruginosa ranked second, after S. aureus, as the most frequently occurring pathogen in the healthcare facilities and ICU in the report of the National Healthcare Safety Network in the United States of America in 2017 (111). About 25% are resistant to carbapenem, cephalosporins, or fluoroquinolones while 18% of ICU patients have serious MDR strains (111). Patients with chronic diseases or immunodeficiency are mostly at risk. P. aeruginosa increases its infection capacity by toxin secretion and biofilm formation (112). The presence of Type III secretion systems is highly associated with persistence of infection, cell apoptosis, and high morbidity (113).
Current diagnostic strategies prioritize antimicrobial susceptibility characterization based on culture samples to guide appropriate antibiotic selection, therapy, and prognosis. Delayed or inappropriate antibiotic treatment has been linked to significantly poor outcomes (114, 115, 116). The debate over monotherapy versus combination therapy persists, with questions regarding their relative superiority. However, a systematic review by Babich et al. (117) encompassing a multicenter analysis of 1119 patients found no clinical advantage between monotherapy and combination therapy in treating bacteremia. Combination therapy offers the advantage of leveraging established synergistic effects by targeting multiple mechanisms of inhibition (114, 118). Conversely, monotherapy proponents argue that new antibiotics can exert more precise and proven antimicrobial effects, aimed at achieving a cure (114, 118).
Resistance due to β-lactamases is one of the predominant phenotypes in P. aeruginosa which could be avoided with the use of carbapenems or a new generation of β-lactams. On the other hand, expression of carbapenemases, especially metallo- beta-lactamases, cefiderocol can be the safe therapy that is also applicable to P. aeruginosa (114). In 2018, “difficult-to-treat” (DTR) P. aeruginosa was coined to recognize its broad resistance to many antibiotics such as aztreonam, ceftazidime, cefepime, ciprofloxacin, imipenem-cilastatin, levofloxacin, piperacillin-tazobactam, and meropenem (119). Depending on patients’ medical circumstance, antibiotic choice and regimen can be formulated. The new antibiotic options for those with pyelonephritis and UTI recommended by Infectious Diseases Society of America are as follows: ceftolozane-tazobactam, ceftazidime-avibactam, imipenem-cilastatin-relebactam, and cefiderocol (118). Meanwhile, ceftolozane-tazobactam, ceftazidime- avibactam, and imipenem-cilastatin-relebactam can be utilized for infections other than the urinary tract (118). The use of ceftolozane-tazobactam, a cephalosporin and β-lactamase inhibitor, has already been approved with significantly lower mortality risk compared to meropenem in the case of ventilated hospital-acquired bacterial pneumonia (120). The aforementioned combination is generally administered as a monotherapy to treat complicated UTI, and pyelonephritis with a recently established pediatric safety (118, 121). The pair of ceftazidime-avibactam works under the principle that avibactam deactivates β-lactamases in class A and C and a number of class D while restoring the susceptibility to ceftazidime (122). Lastly, imipenem-cilastatin-relebactam can be utilized for critically ill patients who have hospital-acquired/ventilator- associated bacterial pneumonia as proven by the phase III clinical trial reported by Titov et al. (123).
Phage therapy authorization is gradually transitioning towards clinical trials. A notable example is a phase I/II trial conducted in France, where a cocktail of bacteriophages was used against P. aeruginosa infections in wound burns (124). The study had a small sample size (n=27) demonstrating a relevant decrease in bacterial load but with slower progression with those treated with the phage cocktail than standard-of-care (124). Challenges on creating a standard phage treatment requires a substantial cohort to further evaluate and explore its potential against MDR- and XDR-P. aeruginosa. In a case report of Law et al. (125) concerning a 26-year old patient with cystic fibrosis, successful and safe complementary bacteriophage therapy was demonstrated with no recorded adverse effects and pseudomonal recurrence. Another case report on personalized bacteriophage therapy showed clinically significant advancement as a complementary treatment, together with IV antibiotic regimen, against MDR-P. aeruginosa (126). Phage vFB297 from the genus of Pakpunavirus was administered through a nebulizer for 20 minutes for five consecutive days and cleared any respiratory obstruction shown in CT scan (126). The patient is chronically infected by MDR-P. aeruginosa making eradication close to impossible but the potential for future use of phage therapy against acute cases can be an alternative. However, challenges persist in selecting appropriate phages and strains of MDR-P. aeruginosa, posing obstacles to advancing into clinical trials and ensuring accessibility of such treatments once developed.
MDR-Mycobacterium tuberculosis
Multidrug-resistant tuberculosis (MDR-TB) is characterized by immunity to isoniazid and rifampicin hence requiring a lengthened treatment recommended by the WHO against MDR-M. tuberculosis (127). Recent standardized treatments lowered the 18-24 months treatment regimen by 9-12 months (127). One reatment regimen using delamanid, linezolid, and levofloxacin with the addition of pyrazinamide obtained a promising 9-month treatment period against fluoroquinolone-sensitive MDR-M. tuberculosis in 12 participating hospitals in South Korea (128). Delamanid is verified as safe treatment for adults which has been now demonstrated to be applicable treatment to children upon pediatric formulation (129). However, both linezolid and levofloxacin posed adverse effects in adults and children that must be considered upon regimen selection and formulation, respectively (130, 131). Meanwhile, Esmail et al. (132) showed that the oral regimen of levofloxacin, bedaquiline, and linezolid can shorten the treatment to six (6) months compared to the 9-month standard-of-care treatment by WHO. Risk to benefit ratio must be further elucidated to lessen the burden of observed toxicity as the study only included those 18 years old and above with specific drug susceptibility requirements (132). The use of NPs in treating MDR-TB and extensively drug-resistant TB is still lacking and pioneering. The current knowledge is that AgNPs and zinc oxide showed anti-mycobacterial suppression but not the capacity to kill MDR- and XDR-M. tuberculosis strains (133). Meanwhile, mutation in the katG gene that renders resistance to isoniazid can be further addressed through the carbon nanotube delivery system (134). This works in multi-walled carbon nanotubes where the antibiotic 4-pyridine carboxylic acid hydrazide (INH) and fluoxetine exert effective and additive results to circumvent the present defense mechanisms of MDR-TB and XDR-TB (134). Successful bacterial cell wall destruction and lowered safe doses of fluoxetine to kill TB-infected macrophages were the highlights of the new described delivery system (134). It is imperative to note that further evaluations under animal models up to clinical trials must be pursued.
In summary, the global health-care burden is presently overwhelmed by the narrowing effective antibiotics and limiting treatment strategies. Combination antibiotic therapies and use of novel approved antimicrobial drugs are still the main approach against priority pathogens. Although the WHO encourages the acceleration of development of new generation antibiotics, challenges regarding the lengthy processes until approval, research and development investment and cost, and slim success rates are still the major hindrances faced by the government and private sector. Moreover, the foundational trials for the use of phage therapy and NPs are still limited despite promising in vitro findings and few prospects in the conducted animal research.
EMERGING MOLECULAR STRATEGIES FOR COMBATING ANTIBIOTIC RESISTANCE
Novel strategies are currently being explored to address the increasing difficulty in diagnosing and treating infections in the post-antibiotic era. Some short-term strategies involve various antibiotic combination treatments (135) and antibiotic-free approaches such as phage-based therapies and vaccines. These methods, however, only work selectively on certain bacterial strains, leaving gaps in addressing the full spectrum of potential pathogens (136, 137). While vaccines prove effective as preventive measures, they cannot be used to treat acute infections (138, 139, 140). Furthermore, other approaches like microbiome manipulation or host immune system modulation have shown limited success (22). Consequently, there is an urgent need for new approaches to antimicrobial therapy that can deliver better outcomes. This section provides an overview of emerging strategies for combating antibiotic resistance, including nanomaterials-based antimicrobials and CRISPR/Cas systems integrated with nanomaterial-based delivery.
Nanomaterials-based Antimicrobial Agents
The number of published works on nanomaterials has grown exponentially over the past years (141). A summary of the recent advances in targeted nanomaterials-based antibacterial therapy is already provided by Geng et al. (142). Nanomaterials refer to substances, typically metals, and polymers, that are engineered or manipulated at the nanoscale, which usually range from 1 to 100 nanometers (143). Their therapeutic activity is influenced by their shape, size, and surface chemistry (144). The sizes and structures of various nanomaterials are similar to bacterial biomolecules, enabling diverse interactions with cellular components. This characteristic can be finely tuned through surface modifications, making them formidable tools against antibiotic-resistant pathogens (145). Some next-generation nanomaterials can also exhibit antimicrobial properties when activated by external stimuli. These types of nanomaterials can remain dormant until triggered by certain forms of stimuli such as light, magnetism, ultrasound, or a change in pH. This unique property makes them less likely to cause any harmful side effects on host cells or non-pathogenic microbes (142).
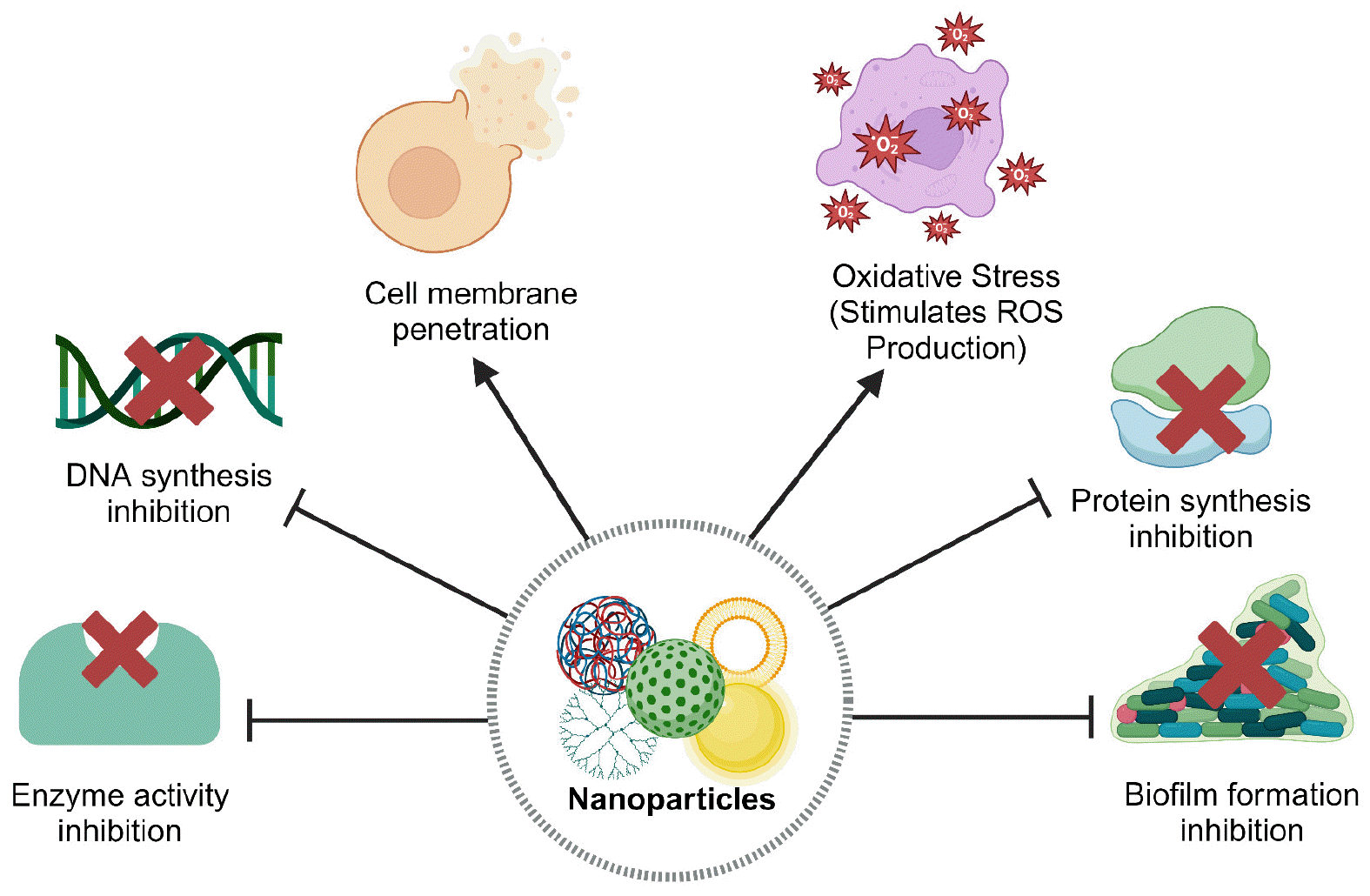
The use of nanomaterials in antimicrobial therapy, as an alternative or complement to existing antibiotics, shows the potential to intervene where conventional antibiotics fail (146). Numerous studies have demonstrated that nanomaterials utilize diverse antibacterial mechanisms, making it harder for pathogens to develop resistance (147). These mechanisms include (1) cell membrane penetration, (2) oxidative stress via the production of reactive oxygen species (ROS), (3) DNA synthesis inhibition, (4) enzyme activity inhibition, (5) protein synthesis inhibition, and (6) prevention of biofilm formation (Fig. 1). The work of Ndayishimiye et al. (148) provides a detailed description of the general types of NPs used for antimicrobial applications (porous silica NPs, liposomes, polymeric NPs, dendritic NPs, metallic NPs) and their mechanisms of action. However, some findings suggest that nanomaterials trigger the evolution of AMR, particularly, metal oxide NPs (149). These conflicting findings were addressed in the review of Xie et al. (150).
Fig. 1
Common antibacterial mechanisms of nanoparticles. Adapted from Karnwal et al. (145). Created with BioRender.com.
NPs could also be functionalized with biomolecules that have an affinity to the external structures of bacteria. This approach greatly enhances the ability of the NPs to selectively interact with the targeted pathogens, thus improving therapeutic outcomes and reducing side effects (151). Gao et al. (152) showed that poly (lactive-co-glycolic acid) (PLGA) NPs preloaded with antibiotics and coated with the membrane of S. aureus-secreted extracellular vesicles could actively target both S. aureus infected macrophages in vitro and heavily infected major organs of mice models, particularly the kidneys and lungs. A “smart” delivery of certain antibiotics in infected tissues may also be done. As demonstrated in the study by Wang et al. (153), moxifloxacin, a broad-spectrum antibiotic, was encapsulated in 4-(hydroxymethyl) phenylboronic acid pinacol ester (HPAP)-modified cyclodextrin (Oxi-αCD). This material rapidly breaks down in the presence of 0.5 millimolar (mM) of hydrogen peroxide enabling the controlled release of the drug to infected lung tissues with abnormally high ROS levels (154, 155). The surface of the NPs was coated with 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-methoxy (polyethylene glycol) (DSPE-PEG) and folic acid-conjugated1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-methoxy (polyethylene glycol)-3400 (DSPE-PEG-FA) to enable targeted elimination of infected macrophages and mucus penetration in the infected areas. Previous studies have shown that Folic-acid-modified NPs can target infected macrophages that overexpress folate receptors (FR) (156, 157). Intravenous delivery of these engineered NPs resulted in an increased survival rate in P. aeruginosa-infected mice. The NPs were also found to have accumulated in infected lung tissues suggesting enhanced active targeting ability.
Out of the five identified NPs commonly used for antimicrobial therapy, metallic NPs have the most direct antimicrobial activity. Metal ions released from the extracellular space can penetrate the cells and disrupt their biological functions, leading to the production of ROS. The resulting oxidative stress also hinders the oxidation of glutathione, which is an important defense mechanism employed by bacteria against ROS (158). Metallic-based NPs can form nonspecific links with organic compounds and other cellular components, which can result in a wide range of effects. For example, these NPs can cause damage to DNA and proteins, which can in turn impact cell division and enzymatic activity (147, 159). Furthermore, metallic NPs can disrupt protein synthesis by causing the denaturation of ribosomes (160). The paper of Sklodowski et al. (161) provides a comprehensive review of the synthesis and design of metallic NPs and metallic NP-based nanosystems (used in combination therapy) for the treatment of drug-resistant bacterial and fungal infections.
For instance, AgNPs are at the forefront of next-generation antibiotics and are the most widely used NPs among all commercialized nanomaterials due to their remarkable antimicrobial properties (162). AgNPs can penetrate bacterial membranes and release Ag+ ions which can trigger the release of ROS, causing oxidative stress. Ag+ ions and AgNPs can also bind to DNA leading to protein inactivation and eventual cell death (163). A recent study evaluated the antimicrobial activity of AgNPs in combination with conventional antibiotics against wild-type Gram-positive and Gram-negative bacteria including antimicrobial-resistant ESBL-positive K. pneumoniae. Results showed that AgNPs exhibited significant antimicrobial activity against Gram-negative wild-type strains of E. coli, A. baumannii, K. pneumoniae, P. aeruginosa, (MIC: 16-128 μg/mL) while minimal to no antimicrobial activity was observed against Staphylococcus saprophyticus, S. aureus, Staphylococcus sciuri, and S. epidermidis. AgNPs were also effective against the antimicrobial-resistant K. pneumoniae isolates. Moreover, AgNPs used in conjugation with kanamycin, colistin, rifampicin, or vancomycin resulted in remarkable synergistic activities against K. pneumoniae clinical isolates. The AgNPs and kanamycin combination demonstrated synergistic antimicrobial activity against all AMR K. pneumoniae clinical isolates (164). These results suggest that the several possible interactions between AgNPs and the different types of antibiotics, and the inherent genetic makeup of isolates may be the cause of variations in susceptibility. It was, however, demonstrated in the study of Lu et al. (165) how exposure of bacteria to sub-lethal concentrations of AgNPs and Ag+ ions accelerated the horizontal gene transfer of antibiotic resistance genes. It was observed that the levels of ROS production, increased membrane permeability, and SOS response of the cell play a role in the enhanced conjugative transfer of resistance genes.
Overall, nanomaterials hold significant potential in combating infectious diseases. They offer numerous advantages, including enhanced solubility, targeted drug delivery, controlled release, and enhanced cellular uptake (166). Table 1 provides a summary of clinical trials that highlight the recent exploration of various nanoparticle technologies for combating bacterial infections. AgNPs are prominently featured in various formulations and applications, targeting a diverse range of pathogens, including antibiotic-resistant strains. Additionally, a significant focus on dental applications is evident, with trials investigating the efficacy of NPs in caries management and oral hygiene (Table 1). While the focus of the presented clinical trials is on dental applications, the abundance of ongoing research underscores the potential of nanomaterials in addressing AMR and improving clinical outcomes across diverse healthcare settings.
Table 1.
Different studies under the clinical trial phase (Retrieved and modified from clinicaltrials.gov and trialsearch. who.int)
CRISPR-Cas and Nanoparticle System-Based Strategy
Several papers have shown that NPs used in conjugation with the CRISPR-Cas system show promise as a novel strategy in combating MDR bacterial infections. CRISPR-Cas9 is the most widely used gene editing tool among CRISPR-Cas Systems (167). The CRISPR-Cas is an adaptive immune system of bacteria and archaea against invading viruses and mobile genetic elements (168). The CRISPR-Cas system comprises three primary components: (a) a leading sequence, (b) an operon containing a cluster of genes encoding Cas proteins, and (c) a CRISPR DNA, a series of repetitive sequences (repeats) separated by non-repetitive sequences (spacers).
The CRISPR-Cas system operates in three stages: adaptation, expression, and interference. During adaptation, the Cas1-Cas2 complex integrates invading DNA into the CRISPR array as a new spacer. In the expression stage, the CRISPR array is transcribed into precursor CRISPR RNAs, which are then processed into mature crRNAs with the assistance of tracrRNA. In the interference stage, the mature crRNAs guide Cas proteins to the target DNA, resulting in enzymatic cleavage and a double-stranded DNA break (169). These mechanisms observed in the CRISPR-Cas system make it an effective method for eliminating foreign genetic elements with high precision
The CRISPR-Cas system can be used to make antibiotic-resistant bacteria sensitive to antibiotics again by removing plasmids that carry genes for antibiotic resistance. This method has been successfully used by Kim et al. (170) to eliminate ESBL-producing E. coli bacteria by targeting a conserved antibiotic resistance gene, identified by analyzing the genomes of over 1000 ESBL mutants. ESBL bacteria are typically resistant to multiple antibiotics and can transfer their antibiotic resistance plasmids to other bacteria through horizontal gene transfer. In addition, the CRISPR-Cas system can be used to selectively eliminate resistant strains from complex bacterial populations. This can be achieved by introducing a plasmid or phage carrying an engineered CRISPR-Cas9 system that targets a unique sequence found only in the resistant strains (171, 172). The potential of CRISPR-Cas9 technology in tackling AMR has been highlighted by Javed et al. (173), with results from several preclinical studies demonstrating its effects. However, based on their review, certain obstacles still need to be overcome, such as improving the system’s efficiency and specificity, creating effective methods of drug delivery, and addressing safety concerns. Moreover, the use of CRISPR-Cas antimicrobials may give rise to new antibiotic-resistant strains of bacteria (174).
Nanotechnology can be used alongside the CRISPR-Cas system to enhance its delivery into target cells. However, delivering the naked genetic elements of CRISPR-Cas into cells can be challenging due to various factors like serum protein adsorption, rapid clearance from the bloodstream and phagocyte uptake, inefficient outcomes due to the inability to specifically target the intended cells, and negative immune system reactions leading to toxic side effects. To overcome these issues, researchers have developed polymeric, lipid, and metal NPs which can serve as gene carriers (175).
In order to perform gene editing using CRISPR-Cas9, it is necessary to package the Cas9 endonuclease and the single guide RNA (sgRNA) specific to the CRISPR system into delivery vehicles. Current methods used to administer the CRISPR-Cas9 system, include: (a) employing a plasmid that encodes both Cas9 and sgRNA, (b) utilizing two distinct plasmids for each (Cas9 and sgRNA), (c) deploying mRNA for Cas9 and gRNA, or (d) utilizing a complex of Cas9 and gRNA (176). However, each of these delivery systems has its limitations. The large size of DNA plasmids makes it difficult to penetrate highly selective cellular and nuclear membranes, which can hinder their effectiveness. While mRNA forms of Cas9 and gRNA may overcome these issues, the unstable nature of mRNA and its sensitivity to RNAses makes it an undesirable method of delivery. Furthermore, while viral vectors are commonly utilized for delivering CRISPR-Cas systems, they also present a few disadvantages which include limited loading and packaging efficiency, narrow host range, and an increased likelihood of causing cancer and triggering an unfavorable immune response (177, 178) (Fig. 2). Notably, nanoparticle-based delivery methods using lipid, polymeric, and gold NPs, of the CRISPR system show promise in addressing the aforementioned limitations by enabling targeted delivery and improving stability (179). Wan et al. (177) provide a thorough review of numerous studies on using tailored/modified NPs for CRISPR-Cas9 delivery. Their paper revealed that for CRISPR delivery, lipid NPs have been the most extensively researched NP systems. Modified lipid NPs were found to improve gene knockout efficiency and increase cellular uptake (180, 181). Recently, a study demonstrated how CRISPR-Cas9 was delivered into wild-type colistin-resistant E. coli using modified single-wall carbon nanotubes (SWCNTs). This SWCNT-CRISPR complex yielded promising results through two approaches. First, the CRISPR-Cas9 system cleaved the targeted mcr-1 gene (colistin resistance gene) and the conductive SWCNTs disrupted the electron transfer chain leading to a shortage of ATP supply (182).
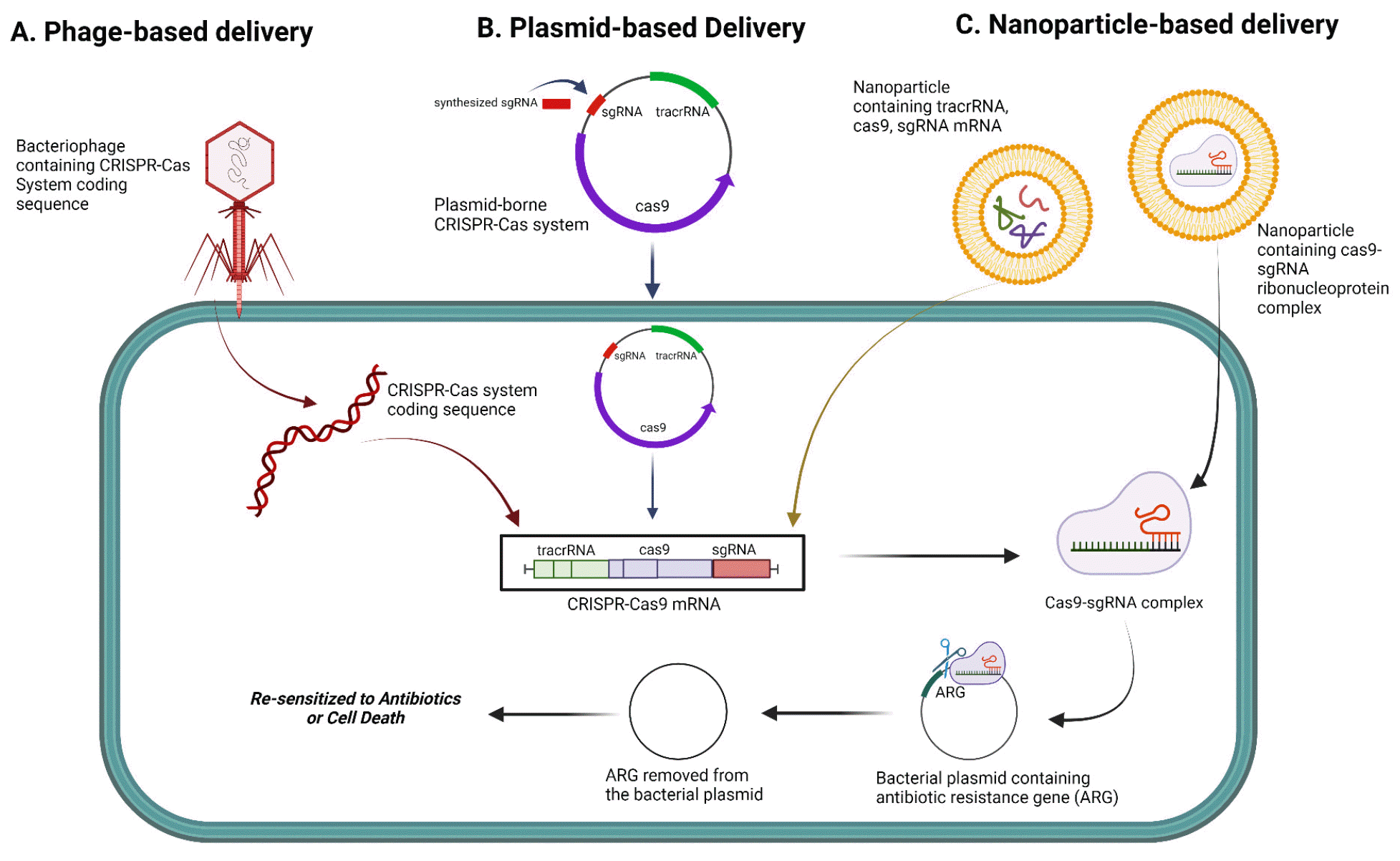
Fig. 2
Delivery methods of the CRISPR-Cas9 system to facilitate gene editing. A. Phage-based delivery: The CRISPR-Cas system sequences are introduced into the cell via phages. B. Plasmid-based delivery: The plasmid-borne CRISPR-Cas system containing a synthesized single guide RNA that targets the antibiotic resistance gene (ARG) can be transferred into cells and transcribed into Cas9 mRNA, tracrRNA, and sgRNA. Following the translation of Cas9 mRNA into the Cas9 protein, it forms a ribonucleoprotein (RNP) complex with sgRNA. Directed by the sgRNA, the RNP complex then edits the target genes. C. Nanoparticle-based delivery: Cas9 and sgRNA can be delivered via mRNA or Cas9-sgRNA RNP complexes, facilitated by NPs. When delivered to bacteria cells the CRISPR-Cas system can eliminate ARG on plasmids and re-sensitize them to antibiotics. Adapted from Tao et al. (178). Created with BioRender.com.
The work of Li et al. (183) demonstrates how CRISPR-Cas systems, NPs, and conventional antibiotics can be used in synergy to address AMR. In their paper, they describe the development and mechanisms of a multifunctional pH-sensitive CRISPR-Cas9-based nanosystem likened to a “nanobomb” against carbapenem-resistant A. baumannii (CRAB). Briefly, CRISPR-Cas9-sgRNA targeted the blaNDM gene to inhibit the translation of metallo-β-lactamases, subsequently re-sensitizing CRAB to imipenem and enhancing the synergistic antimicrobial activities of imipenem and ZnO-SiO2 NPs. Furthermore, L-arginine mixed in the nanosystem reacts with the high ROS levels to form nitric oxide (NO) reaction products which are known antibacterial agents. Conversely, NO also possesses anti-inflammatory properties, promotes proliferation, and accelerates wound healing in infected areas (184). This CRISPR-Cas9 nanosystem also showed promising effects in acute pneumonia and peritonitis mouse models including mitigating infections, inhibiting inflammatory reactions, and promoting tissue repair of infection sites (183). Combining engineered NPs/nanosystems and CRISPR-Cas systems holds great potential in tackling AMR. However, this field of research is still in its early stages and requires further exploration and development.
CONCLUSION
AMR poses a significant and growing global health threat, jeopardizing the effectiveness of existing antibiotics and highlighting the urgent need for novel therapeutic strategies. This work has comprehensively explored the complexities of AMR, encompassing its emergence, mechanisms, and detrimental impact. While the discovery and development of new antibiotics remain crucial, this review emphasizes the promise of emerging molecular approaches, namely nanomaterials- based antimicrobials and CRISPR-Cas systems, in combating AMR. However, further research and development are essential to refine these strategies, address limitations such as cost-effectiveness and safety concerns, and ensure their successful translation into clinical practice.
 XML Download
XML Download