

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
MAIN BODY
1. Complement pathways
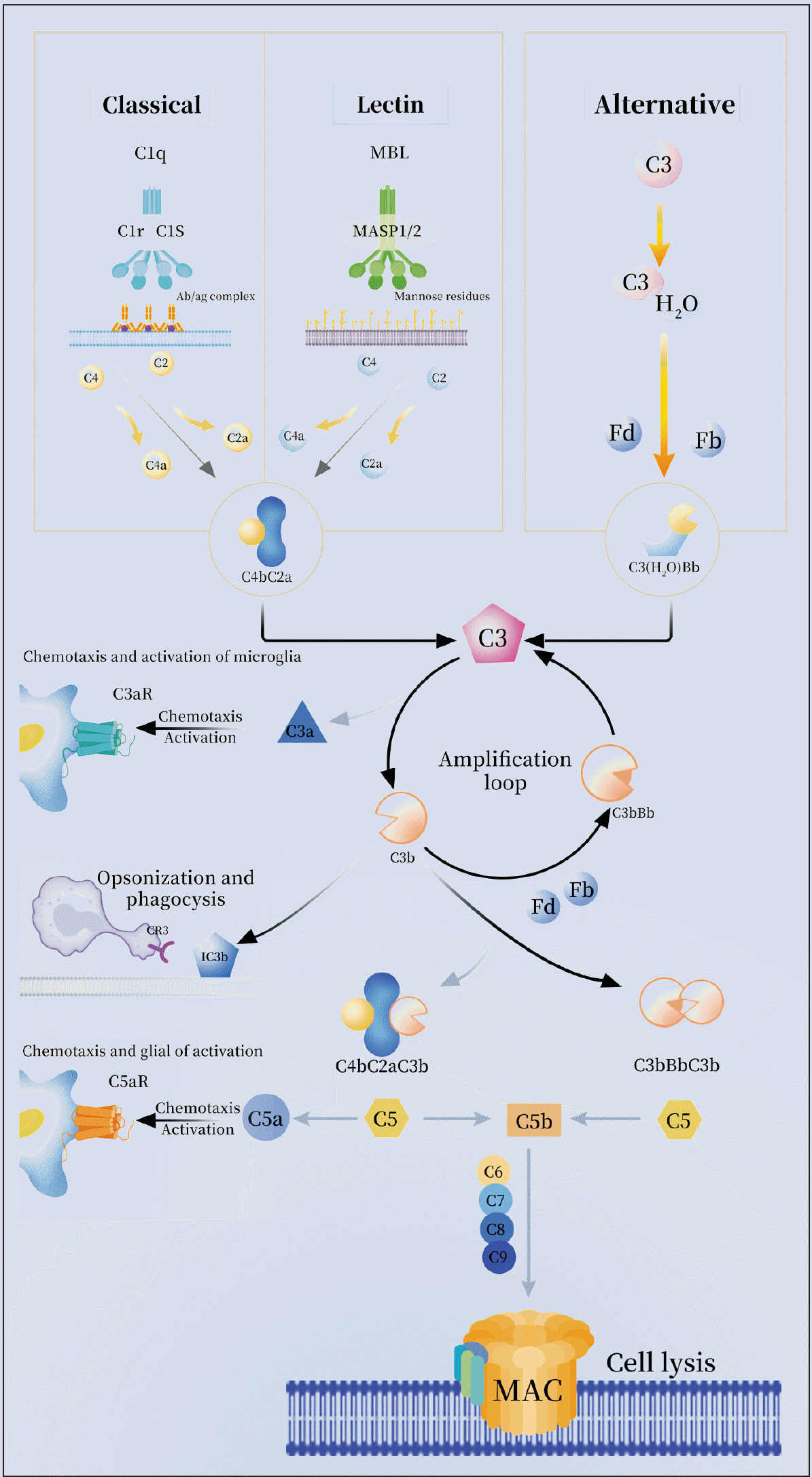 | Fig. 1Activation pathways of the complement system. The complement system is activated by the classical, lectin, or alternative pathways. The initiation of the classical pathway involves the C1 protein complex, which consists of C1q, C1r, and C1s. Upon activation, C1s cleaves C4 and C2. In the lectin pathway, activation occurs when mannose-binding lectin (MBL) encounters a pathogenic carbohydrate motif. Subsequently, MBL associates with MASP-1 and MASP-2, leading to the cleavage of C4 and C2. The resulting cleavage products induce the formation of C3 convertase C4bC2a, which subsequently splits C3 into C3a and C3b. C3a has the ability to induce the chemotaxis and activation of microglia via C3aR. Additionally, C3b can undergo cleavage to form iC3b, which is recognized by microglial cells through complement receptor 3 (CR3), thereby promoting activation. Furthermore, the production of C3b can facilitate the generation of C5 convertase (C4bC2aC3b, C3bBbC3b). C5 can also be cleaved into C5a and C5b, with C5a further promoting the chemotaxis and activation of glial cells through C5aR. Another pathway involves the spontaneous hydrolysis of C3 to C3 (H2O). All three pathways result in the generation of convertases, which subsequently stimulate the production of the primary components of the complement system, namely anaphylatoxins (C4a, C3a, and C5a), membrane attack complexes, and opsonins (C3b). Allergic toxins initiate pro-inflammatory signaling, while C5b associates with C6, C7, C8, and C9 to form membrane attack complexes (MAC), ultimately resulting in cell cleavage.
|
2. Complement system in chronic pain and depression
3. The role of complement system-mediated glial cell synaptic pruning in the comorbidity of chronic pain and depression
1) Synaptic pruning by microglia and astrocytes
2) Complement system participated in glial cell-mediated synaptic pruning
3) C1q/C3-CR3 in chronic pain and depression comorbidity
Table 1
| Complement cascade | Molecular mechanisms | Method/experimental models | Type of disease |
|---|---|---|---|
| C1q | C1q is the hub gene of NP [14] | SNI model, random walk with restart (RWR) methodology [14] | NP |
| C1q | The knockout of C1q led to a significant increase in the mRNA expression of pro-inflammatory cytokines TNF-α, IL-6, and IL-1β in the PFC of mice [68] | The LH model of depression [68] | Depression |
| C3 | C3 is the hub gene of NP [13,14] | SNI model, RWR [13,14] | NP |
| C3 | The expression of PFC C3 increased significantly in depressed suicide patients. Chronic stress leads to increased C3 expression in PFC [20] | CUMS mice [20] | Depression |
| C1q/C3 | Neuroinflammation was observed in the glial cells, of the amygdala, C1q and C3 activation, Syn and PSD-95 levels decreased [48] | CRS mice [48] | Chronic pain and depression |
| C1q/C3-CR3 | BoNT/A treatment decreased the levels of complement C3\CR3 and C1q, decreased the colocalization of iba-1 and CD68, decrease the mRNA expression of CX3CR1 in microglia cells, recovered the density of PSD-95, decreased the mRNA levels of TNF-α and IL-1β [49] | PD mouse model, Mice were administered reserpine (3 μg/mL in the drinking water) for 10 wk. BoNT/A (10 U·kg-1·d-1) was injected into the cheek for 3 consecutive days [49] | Chronic pain and depression |
| C1q/C3-CR3 | The mRNA levels of amygdala complement C1q and ITGAM exhibited a significant increase in microglia within the CeA. Iba-1 and PSD95 colocalization levels increased [50] | Fischer-344 rats, micropellets containing either corticosterone (CORT) were bilaterally implanted onto the CeA using stereotaxic techniques [50] | Chronic pain and depression |
| C3/C3a-C3aR | Treatment with hUC-MSCs and a C3aR antagonist resulted in an increase in protein levels of PSD-95 and AMPA, suppressing TNF-β and IL-10. CD16+/Iba1+ in the hippocampus and the level of C3a protein in GFAP+ cells were inhibited by hUC-MSCs [53] | CUMS mice, hUC-MSCs were administered intravenously to CUMS mice once a week for a duration of 4 wk [53] | Chronic pain and depression |
| C3/C3a-C3aR | LPS resulted in the activation of the C3/C3aR in PFC increased polarization of microglia, hUC-MSCs can reduced the C3aR and STAT3, C3aR antagonism treatment restored PSD-95 and SYN levels and improved microglia morphology IL-1R blockers block activation of the pNF-κB/C3 pathway in neurotoxic A1 astrocytes [25] | CUMS mice, hUC-MSCs mice was injected with LPS (0.83 mg/kg, i.p.), The animals received intraperitoneal injections of either saline or C3aR blockade once a day for 4 wk [25] | Chronic pain and depression |
| C3/C3a-C3aR | Gypenoside XVII: resulted in a decrease in the number of Iba1 positive cells and an upregulation of C3 levels, the downregulation of pSTAT3, the levels of IL-1β, IL-6 and TNF-α in the PFC, reduction of VGIut2 within the microglial and PSD95 [54] | CUMS mice, Gypenoside XVII was dissolved in a solution of 0.9% saline containing 0.3% carboxymethyl cellulose and administered orally once daily for a duration of 4 consecutive wk [54] | Chronic pain and depression |
| C3/C3a-C3aR | The downregulation of C3aR was observed to inhibit the activation, of A1 astrocytes induced by LPS, resulting in a decrease in the expression of C3aR, C3, and GFAP. These proteins are known to be involved in the transition from acute to chronic pain [57] | The induction of A1 astrocytes was achieved through intraperitoneal injection of LPS [57] | Chronic pain and depression |
NP: neuropathic pain, SNI: spared nerve injury, TNF-α: tumor necrosis factor-α, IL-1β: interleukin-1β, PFC: prefrontal cortex, LH: learned helplessness, CUMS: chronic unpredictable mild stress, PSD-95: postsynaptic density protein 95, CRS: chronic restraint stress, SYN: synaptophysin, BoNT/A: botulinum neurotoxin A, CX3CR1: C-X3-C motif chemokine receptor 1, PD: Parkinson’s disease, ITGAM: integrin, alpha M, CeA: central nucleus of amygdala, hUC-MSCs: human umbilical cord mesenchymal stem cells, LPS: lipopolysaccharide, STAT3: signal transducer and activator of transcription 3, NF-κB: nuclear factor-kappa B, VGlut2: vesicular glutamate transporter-2.
![]()
4) C3/C3a-C3aR in chronic pain and depression comorbidity
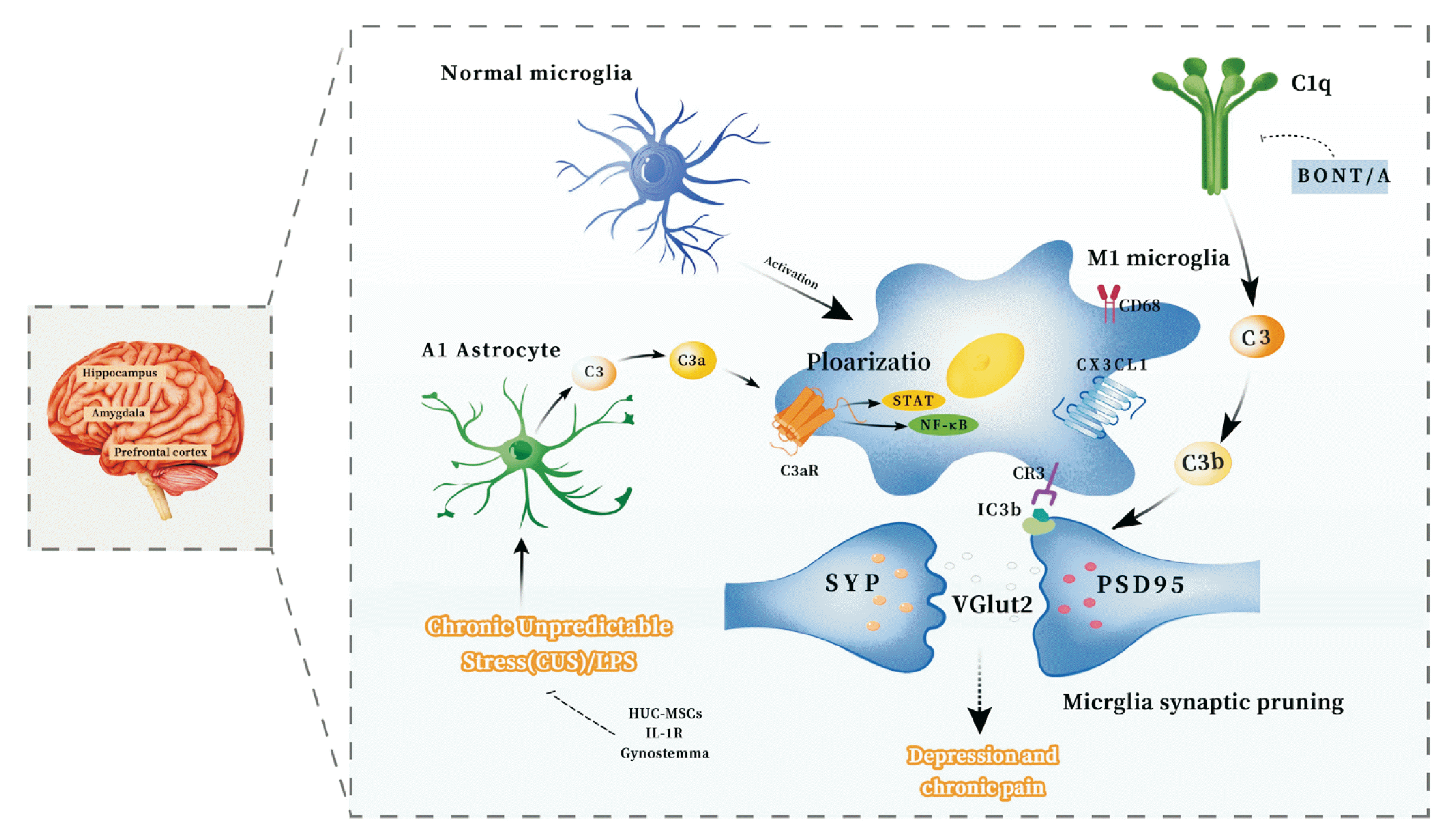 | Fig. 2Potential mechanisms by glial cell synaptic pruning mediated by the complement system in chronic pain and depression comorbidity. Complement cascade C1q/C3-CR3 pathway. The amygdala and hippocampus exhibit heightened activation of complement C1q and C3 in response to stimuli associated with chronic pain and depression. This activation facilitates the differentiation of microglial cells into a pro-inflammatory M1 phenotype. Complement C3 undergoes lysis, leading to the generation of C3b and iC3b. This iC3b acts as a marker for synapses, facilitating their identification by the CR3 receptor on pro-inflammatory M1 microglial cells, potentially resulting in synaptic pruning. Notably, there is a reduction in the levels of synaptic proteins SYP and PSD95, as well as a decrease in the secretion of the VGlut2. The BonT/A can counteract these changes by inhibiting the activation of C1q and C3, thereby reducing synaptic pruning in microglia. Complement cascade C3/C3a-C3aR signals pathway. In the presence of LPS or chronic stress, neurotoxic A1 astrocytes in the prefrontal cortex become activated, resulting in an increase in complement C3 and C3a levels. Specifically, C3a targets the C3aR receptor on microglial cells, promoting their polarization towards an M1 phenotype. This activation further stimulates the STAT3 and NF-κB pathways in microglial cells. This amplifies synaptic pruning in microglia and hUC-MSCs, IL-1R, and Gynostemma inhibit and improve these processes. PSD95: postsynaptic density protein 95, VGlut2: vesicular glutamate transporter-2, BoNT/A: botulinum neurotoxin A, LPS: lipopolysaccharide, STAT3: signal transducer and activator of transcription 3, NF-κB: nuclear factor-kappa B, hUC-MSCs: human umbilical cord mesenchymal stem cells.
|
4. The complement system mediates glial neuroinflammation and plays a role in the chronic pain and depression comorbidity
1) Relationship between neuroinflammation and chronic pain and depression
2) Complement system-mediated glial cell induced neuroinflammation
3) C3-C3aR axis and chronic pain and depression comorbidity
4) C5-C5aR axis and chronic pain and depression comorbidity
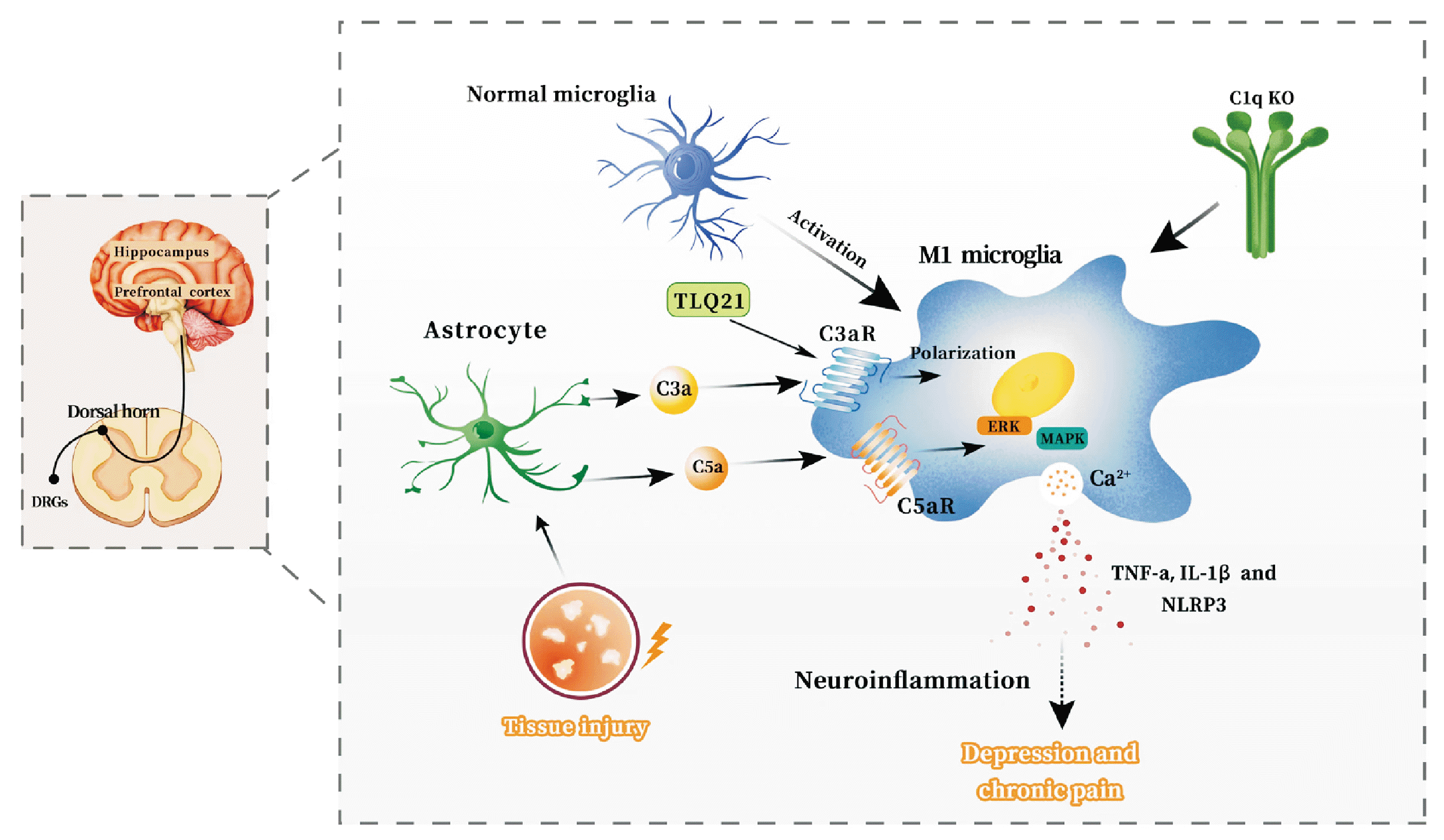 | Fig. 3Potential mechanisms in which neuroinflammation is mediated by the complement system is involved in the comorbidity of chronic pain and depression. Complement cascade C3a-C3aR pathway. Under the influence of injury and inflammation, astrocytes have the ability to enhance the production of complement C3a. This molecule then interacts with the receptor C3aR found on microglia, resulting in the promotion of M1 type polarization of microglia in specific regions of the central nervous system, including the dorsal horn of the spinal cord, dorsal root neurons in the spinal cord, hippocampus, and prefrontal cortex. Consequently, this process leads to an upregulation of the expression of pro-inflammatory cytokines such as TNF-α, IL-1β, and NLRP3 in the hippocampus, as well as an elevation in the levels of the inflammatory cytokine IL-1β in the prefrontal cortex. These events ultimately induce neuroinflammation, which is closely associated with the development of chronic pain and depression. Complement cascade C5a-C5aR pathway. Response to injury and inflammation, astrocytes have the capacity to elevate the expression of complement C5a. The interaction between C5a and its receptor (C5aR) can subsequently enhance the secretion of pro-inflammatory factors via the activation of p38MAPK and ERK1/2 signaling pathways. This process further facilitates the M1-type polarization of microglia in the spinal dorsal horn and dorsal root ganglion (DRG), thereby inducing neuroinflammation. Ultimately, these events contribute to the development of chronic pain and depression. TNF-α: tumor necrosis factor-α, IL-1β: interleukin-1, NLRP3: NOD-like receptor protein 3.
|
Table 2
| Complement cascade | Molecular mechanisms | Method/experimental models | Type of disease |
|---|---|---|---|
| C5/C5aR | Peripheral complement component C5 and C5aR is upregulated in spinal microglia after peripheral nerve injury [23] | SNI, CCI and SNL model [23] | Neuropathic pain |
| C5a-C5aR | The levels of C5a and C5 in plasma and cerebrospinal fluid are significantly increased in patients with major depression [11,77,78] | Depression | |
| C3a-C3aR | C3a can enhance calcium current and capsaicin-induced calcium inflow in dorsal root neurons of spinal cord [23]. TLQP-21 activates the C3aR1 receptor on the dorsal horn microglia of the spinal cord, which induced heat hyperalgesia and contributed to nerve injury-induced [73] |
SNI, CCI and SNL model [23] SNI model [73] |
Chronic pain and depression |
| C3a-C3aR | C3a-C3aR signaling pathway promotes M1 polarization of microglia and increases levels of hippocampal pro-inflammatory cytokines TNF-α and IL-1β [53]. C3aR blockade decreased the expression and inflammatory response of NLRP3 inflammasome in the hippocampus [74] C3a-C3aR monocytes infiltrated the prefrontal cortex, and the levels of inflammatory cytokine IL-1β in the PFC increased [20] | CUMS mice [20,53,74] | Chronic pain and depression |
| C5a-C5aR | C5a can induce upregulation of pro-inflammatory factors such as IL-6, TNF-α depending on C5aR [75]. C5a mediates neuropathic pain by recruiting macrophages and T cells through C5aR. C5a in DRGs may cause direct sensitization of primary sensory neurons [26] | Transgenic MAFIA mice drug-inducible macrophage depletion [76] | Chronic pain and depression |
SNI: spared nerve injury, CCI: chronic constriction injury, SNL: spinal nerve ligation, CUMS: chronic unpredictable mild stress, TLQP-21: TLQP-21 Trifluoroacetate, TNF-α: tumor necrosis factor-α, IL-1β: interleukin-1, NLRP3: NOD-like receptor protein 3, PFC: prefrontal cortex, DRG: dorsal root ganglia, MAFIA: macrophage Fas-induced apoptosis.
![]()
 XML Download
XML Download