

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Congenital factor V (FV) deficiency (FVD) is caused by variants in the F5 gene on chromosome 1q24.2 and accounts for 8% of rare bleeding disorders [1]. Clinical FVD manifestations range from asymptomatic to life-threatening bleeding, making diagnosis and prediction difficult. FVD is clinically suspected when there is a prolonged prothrombin time (PT) and activated partial thromboplastin time (aPTT), which are corrected in the mixing test and confirmed by reduced FV activity. In this report, we describe a patient with hereditary FVD and compound heterozygous variants, including a novel pathogenic variant.
CASE REPORT
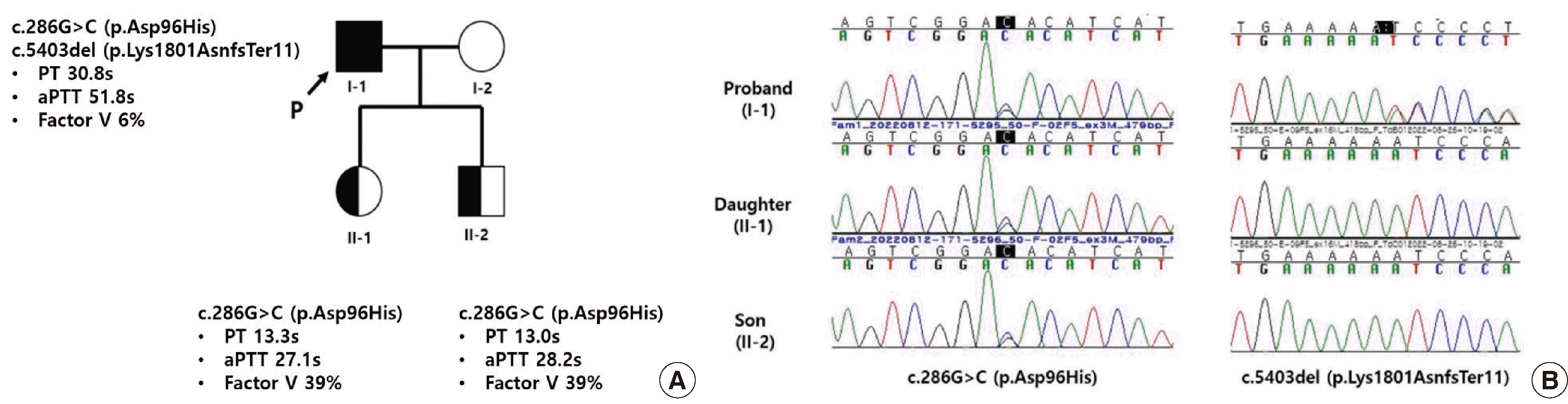
A 50-year-old male was referred to the hematology clinic because of prolonged PT and aPTT during a preoperative examination. He had a right parotid mass that needed excision. The patient had no medical or family history of bleeding. Prolonged PT (30.8 s; reference range, 10.8–14.0) and aPTT (51.8 s; reference range, 21.0–34.0) were corrected in the mixing test, and reduced FV activity (6%) was confirmed. Factor II and VII-XII assays showed no specific findings. Considering the discrepancy between the patient’s clinical symptoms and FV activity, as well as the presence of two children in his family tree, genetic studies and counseling were conducted.
Genomic DNA was extracted from the peripheral blood of the patient and his two children. A 108-gene panel by next-generation sequencing (NGS) for inherited coagulation disorders revealed that the patient had a compound heterozygous variant in the F5 gene (NM_000130.5): a missense variant c.286G>C (p.Asp96His) and a frameshift variant c.5403del (p.Lys1801AsnfsTer11), which is a novel variant. As this novel variant has not been reported before, we deposited it in the ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar/; September 10, 2022) [2]. The variant was classified as pathogenic according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) guidelines: frame-shift null variant in the F5 gene for which loss-of-function is a known mechanism of disease (PVS1); not found in genome AD exomes of the Korean Reference Genome Database (PM2); International Society on Thrombosis and Hemostasis (ISTH) committee recently reports the variant as pathogenic (PP5); patient’s phenotype or family history is highly specific for a disease with a single genetic etiology (PP4). We further confirmed that the patient’s two children inherited the heterozygous c.286G>C (p.Asn96His) variant (Fig. 1). The patient’s FVD was confirmed through genetic study, and the biopsy performed simultaneously showed that the mass was a Warthin tumor with no possibility of malignancy, so the surgery was cancelled. Written informed consent was obtained from the patient for the publication of this case report.
DISCUSSION
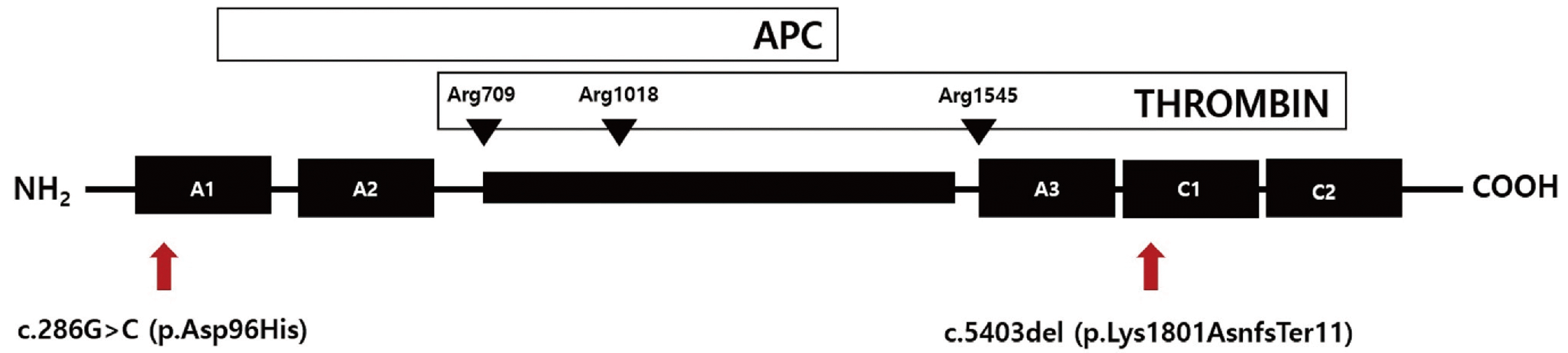
The F5 gene consists of six domains: A1, A2, B, A3, C1, and C2. To date, 265 variants in F5 have been reported, mainly missense variants with high clustering in the A2 and C2 domains (Human Gene Mutation Database professional version (https://www.nihlibrary.nih.gov/resouces/tools/human-gene-mutation-databaseprofessional), last accessed on July 10, 2023). The c.286G>C (p.Asp96His) variant is located in the A1 domain and affects protein folding, thereby interfering with intracellular trafficking and secretion. This variant was previously described in four unrelated Chinese patients with FVD [3]. Since then, three cases have been reported in Korea, two of which were related (Table 1) [1, 4].
The patient in this case was a compound heterozygote with not only the c.286G>C (p.Asp96His) variant but also a novel variant, and therefore had a significantly lower FV activity than previously reported patients with only the c.286G>C (p.Asp96His) variant. This novel variant is located in the C1 domain of the FV protein, which is a mutational hot spot that is predicted to create a frameshift that results in a premature stop codon because of a 1-base pair deletion (Fig. 2), thus changing the protein length. The exact impact of this novel variant cannot be perfectly predicted; however, previously reported cases of compound heterozygous variants, including c.286G>C (p.Asp96His), can explain the very low FV activity observed in this patient [1, 4].
In rare bleeding disorders, the correlation between clinical severity and coagulation factor activity can vary [5]. Recent studies have highlighted the presence of FV in the platelet compartment, which may contribute to this diverse correlation [6]. In the present case, the patient exhibited very low FV activity but had no bleeding tendencies throughout his life; the deficiency was detected only during the preoperative examination. However, It is important to note that patients with FVD typically experience bleeding after invasive procedures. Therefore, without comprehensive genetic evaluation before surgery, there is a risk of massive bleeding. Furthermore, a familial genetic study confirmed that the patient’s children had inherited the variant, suggesting a potential bleeding tendency.
In conclusion, we present a case of FVD in a patient with no history of bleeding. The novel variant c.5403del (p.Lys1801AsnfsTer11) was identified through genetic testing. We suggest that this variant should be classified as a pathogenic variant. Additionally, this case emphasizes the clinical utility of genetic testing in FVD. The patient in this case was diagnosed with FVD through molecular genetic testing using an NGS-based 108-gene panel. Therefore, genetic testing might be an essential method for diagnosing patients with atypical presentation, particularly when there is a weak association between genotype and phenotype, as seen in FVD.
 XML Download
XML Download