

 PDF
PDF Citation
Citation Print
Print



서 론
2020년 5월 1일 체외진단의료기기법이 시행되면서 체외진단의료기기(in vitro diagnostic medical device, IVD)는 일반의료기기(medical device)와 별도의 관리법으로 관리하기 시작하였다[1]. 체외진단의료기기는 인체 유래물을 체외에서 검사하기 위한 의료기기로 인체에 사용되는 일반의료기기와는 구별되며, 코로나19와 같은 신종 감염병 분야, 유전자 검사를 이용한 개인 맞춤형 정밀의료 분야 등에서도 중요한 역할을 할 것으로 전망된다. 이러한 특성을 반영한 체외진단의료기기법의 시행을 통해 체외진단의료기기의 안전성을 확보하고 품질 향상 등의 발전을 도모할 수 있게 되었다. 체외진단의료기기법은 개인 또는 공중보건에 미치는 잠재적 위험을 고려하여 체외진단의료기기의 등급을 4개의 등급(1–4등급)으로 분류하며, 등급 또는 품목에 따라 식품의약품안전처(Ministry of Food and Drug Safety, MFDS)의 신고(notification), 인증(certification), 혹은 허가(approval)의 대상으로 나누어진다[2]. 허가를 받기 위해서는 반드시 임상적 성능시험(clinical performance study) 자료를 제출하여야 하며, 임상적 성능시험은 인체를 직접 대상으로 하지 않고 인체에서 유래한 검체를 대상으로 하기에 임상시험과는 구분된다.
유럽 연합에서도 2022년 5월 26일부터 기존의 체외진단의료기기지침(in vitro diagnosis medical device directive, IVDD)보다 강화된 체외진단의료기기규정(in vitro diagnostic medical device regulation, IVDR) 제도가 시행되었다[3]. 유럽 체외진단의료기기규정 또한 개인 또는 공중보건에 미치는 잠재적 위험에 따라 4개의 등급(class A–D)으로 나누어지며 임상적 성능시험과 같은 임상적 성능을 입증할 수 있는 자료를 제출하도록 하고 있다. 자가선언(self declaration)이 가능한 일부 class A를 제외한 나머지 체외진단의료기기는 인증기관(notified body)의 심사(review)를 통해 인증을 받아야 한다. 이러한 과정을 거쳐 일정 기준을 충족한 제품만이 Conformité Européenne (CE) 마크를 사용할 수 있고, 유럽 내에서 사용이 가능하다. 기존 체외진단의료기기지침에 따라 CE 마크를 사용해오던 체외진단의료기기(레거시 기기, legacy device)라도 일정 유예기간이 끝나기 전에 강화된 체외진단의료기기규정에 따라 변경(upgrade)을 진행하여야만 유럽에서 지속적인 판매가 가능하다. 의료기기조정그룹(Medical Device Coordination Group, MDCG)은 2022년 5월 레거시 기기 변경을 위한 규정을 추가로 발표하였다[4].
본 연구에서는 저자들이 직접 경험한 체외진단의료기기의 임상적 성능시험과 함께 국내 식품의약품안전처 허가와 유럽 체외진단의료기기규정에 근거한 인증 과정을 공유하여 국내 및 유럽 체외진단의료기기 허가(인증)를 준비하는 연구자와 기업체 담당자에게 실질적인 도움을 주고자 하였다.
재료 및 방법
2. 연구대상
실시간 중합효소연쇄반응법(Real Time PCR)을 이용하여 Mycobacterium tuberculosis (MTB) 및 non tuberculosis mycobacteria (NTM) 감염 진단에 도움을 주는 NextGene MTB/NTM Detection Kit (EONE BIOTECH Co., Ltd., Incheon, Korea)와 human herpes simplex virus 1, human herpes simplex virus 2, Gardnerella vaginalis, Treponema pallidum, Candida albicans 및 Ureaplasma parvum에 의한 성매개 감염 진단에 도움을 주는 NextGene STI 12 Detection Kit 3&4 (EONE BIOTECH Co.) 두 가지 유전자검사시약의 체외진단의료기기 국내 및 유럽 허가(인증)를 진행하였다.
결 과
국내 및 유럽 허가(인증) 과정은 Fig. 1에 정리되어 있다.
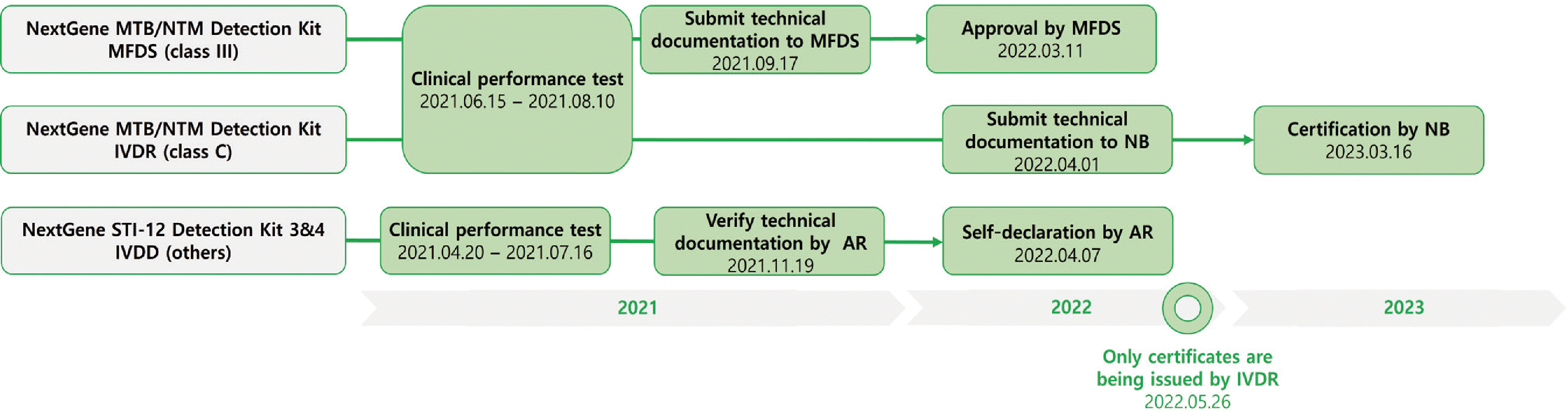
Fig. 1
Timeline of domestic (MFDS) and European (IVDR and IVDD) approval.
Abbreviations: MTB/NTM, mycobacterium tuberculosis/non tuberculosis mycobacteria; MFDS, Ministry of Food and Drug Safety; IVDR, in vitro diagnostic medical device Regulation; NB, notified body; STI, sexually transmitted infection; IVDD, in vitro diagnosis medical device directive; AR, authorized representative.
![]()
1. NextGene MTB/NTM Detection Kit 국내 허가
1) 임상적 성능시험
이원바이오텍에서 가천대학교 길병원으로 임상적 성능시험을의뢰하여 가천대학교 길병원 기관윤리위원회의 승인(GFIRB2021 219)을 받고, 2021년 6월 15일부터 8월 10일까지 진행하였다. 검체는 이원의료재단 기관윤리위원회의 잔여 검체 제공을 위한 승인(128477 202106 BR134)을 얻어 이원의료재단으로부터 객담과 항산균배양 잔여 검체를 제공받아 평가에 이용하였다. 임상적 성능시험의 검체수 산정을 위해 참고문헌 및 기허가 제품의 성능자료를 참조하여 기기의 추정민감도(또는 특이도, P1)와 임상적 목표민감도(또는 특이도, P0)를 설정하였다. 검체수 산정은 다음의 공식으로 계산하였다. n=[(Zα/2+Zβ)2 ×P1(1-P1)]/(P1-P0)2. Zα/2 값은 유의수준을 0.05로 설정하였을 때 1.96, Zβ 값은 검정력을 90%로 하였을 때 1.24였다. 연구 중 누락되는 검체를 고려하여 계산된 n값의 10%를 더하여 검체수를 산정하였다. 기기의 추정민감도(95%)와 임상적 목표민감도(87%)를 이용하여 최종적으로 양성 검체수를 86개로 산정하였고, 기기의 추정특이도(94%)와 임상적 목표특이도(86%)를 이용하여 음성 검체수를 86개로 산정하였다(Table 1). 냉장 및 실온에 24시간 이상 보관되어 안정성을 확인할 수 없는 검체 및 보관량이 0.5 mL 미만인 검체는 시험 대상에서 제외하였다. 확진검사 결과와 비교하여 임상적 민감도와 특이도는 모두 100%를 보였다. 또한 기존에 허가된 AdvanSure TB/NTM real time PCR (LG Lifescience, Seoul, Korea) (체외 제허 13-52호)과 비교한 상관성 평가에서 양성 일치도와 음성 일치도 모두 100%, 카파계수 1을 보여 NextGene MTB/NTM Detection Kit가 기존 허가 제품과 동등한 성능을 보이는 것을 확인하였다(Table 1).
Table 1
Results of agreement between the NextGene MTB/NTM Detection Kit and the AdvanSure TB/NTM real time PCR in sputum and mycobacterial culture specimens
![]()
2) 식품의약품안전처(MFDS) 허가
2021년 9월 17일 NextGene MTB/NTM Detection Kit는 고위험성 감염체 유전자 검사시약 품목에 해당하여 식품의약품안전처 3등급 허가를 신청하였다. 신청 시 제품의 성능을 확인하기 위해 가천대학교 길병원에서 시행한 임상적 성능시험 자료 외에 분석적 성능시험 자료, 품질관리 시험에 관한 자료, 표준물질 및 검체 보관 등에 관한 자료를 함께 제출하였다. 또한 기원·개발경위, 측정 원리·방법에 관한 자료, 국내·외 사용현황에 관한 자료, 원재료 및 제조방법에 관한 자료, 사용목적에 관한 자료, 저장방법과 사용기간 또는 유효기간에 관한 자료, 취급자 안전에 관한 자료 등을 함께 제출하였다[2]. 이후 2021년 12월 16일 1차 보완자료 제출, 2022년 1월 24일 2차 보완자료를 추가로 제출하여 2022년 3월 11일 최종적으로 품목 허가(체외 제허 22-159호)를 받았다.
2. NextGene MTB/NTM Detection Kit 유럽 인증(2022년 5월 26일 이후)
2) 체외진단의료기기규정(IVDR) 인증
2022년 4월 1일 NextGene MTB/NTM Detection Kit는 유럽 체외진단의료기기규정의 분류 규칙(classification rules)에 따라 class C 인증을 신청하였다[5]. 유럽 내 국가별 보건당국의 허가를 받은 인증기관에서 체외진단의료기기규정 부속서 IX (Annex IX)에 따라 제품의 적합성 평가(conformity assessment)를 진행하였다. 적합성 평가를 위해 인증기관은 임상적 성능(clinical performance), 분석적 성능(analytical performance), 과학적 타당성(scientific validity) 등을 포함한 여러 기술문서(technical documentation) 자료와 품질경영시스템(quality management system)을 심사하였다. 2022년 7월 체크리스트 리뷰 및 2022년 12월 서류 보완 과정을 거쳐 2023년 3월 16일 최종적으로 인증(IVDR 763366)을 획득하였다.
3. NextGene STI 12 Detection Kit 3&4 유럽 자가선언 (2022년 5월 26일 이전)
1) 임상적 성능시험
2021년 4월 20일부터 7월 16일까지 가천대학교 길병원에서 기관윤리위원회의 승인(GFIRB2021-139)을 받고 진행하였다. 이원의료재단 기관윤리위원회의 잔여 검체 제공을 위한 승인(128477-202103-BR122)을 얻어 이원의료재단으로부터 소변과 질도말 잔여 검체를 제공받았다. 검체수 산정은 NextGene MTB/NTM Detection Kit와 같은 공식을 사용하였고 각 원인균별로 참고문헌 및 기허가 제품의 성능자료를 참조하여 각각의 민감도와 특이도를 설정하였다. 냉장 및 실온에 24시간 이상 보관되어 안정성을 확인할 수 없는 검체 및 보관량이 0.5 mL 미만인 검체는 시험 대상에서 제외하였다. 다중감염 검체의 경우 4개 이하의 다중감염 검체만을 유효한 검체로 선정하고, 해당 감염 원인균 각각을 양성 검체로 사용하였다. 확진검사와 비교를 통하여 임상적 민감도와 특이도를 평가하였다. 또한 기존에 허가된 PANA RealTyper STD Kit (PANAGENE, Daejeon, Korea) (체외 제허 17-456호)와 상관성 평가를 진행하였다. Candida albicans를 제외한 human herpes simplex virus 1, human herpes simplex virus 2, Gardnerella vaginalis와 Ureaplasma parvum은 확진검사와 비교하여 100%의 임상적 민감도 및 특이도를 보였으며, 기존 허가 제품과 100% 양성 일치도와 음성 일치도를 보였다. Candida albicans의 경우 질도말 검체에서는 2개의 양성 검체(임상적 민감도 97.7%, 양성 일치도 97.7%, kappa 계수 0.98), 소변 검체에서는 3개의 양성 검체(임상적 민감도 96.3%, 양성 일치도 96.3%, kappa 계수 0.97)를 검출하지 못하였으나 모두 유효성 평가 기준을 만족하였다(Table 2).
Table 2
Results of agreement between the NextGene STI 12 Detection Kit 3&4 and the PANA RealTyper STD Kit in vaginal swab and urine specimens
![]()
2) 체외진단의료기기지침(IVDD) 자가선언
NextGene STI 12 Detection Kit 3&4는 체외진단의료기기규정으로 변경되는 2022년 5월 26일 이전에 유럽 체외진단의료기기지침에 근거하여 자가선언(self declaration)을 통해 others 등급으로 체외진단의료기기 등록을 완료하였다. 2021년 10월 22일 유럽대리인(authorized representative)과 계약 후, 2021년 11월 19일 유럽대리인에게 기술문서 등의 검토를 의뢰하였다. 이후 2022년 4월 7일 최종적으로 유럽대리인에 의해 자가선언이 완료되었다. 또한 국내 체외진단의료기기법은 해외판매 제품에 대해 수출용 허가를 요구하고 있어 2022년 8월 3일 수출용 허가를 신청하여 같은 해 8월 16일 식품의약품안전처 허가(체외 제허 22-554)를 획득하였다.
고 찰
국내 체외진단의료기기의 생산실적은 2020년에 3조 3500억 원, 2021년에 4조 3500억 원으로 매년 증가하고 있으며, 2021년 일반 의료기기 생산실적이 8조 5300억 원인 것과 비교해 보면 전체 의료기기에서 체외진단의료기기의 비중이 큰 것을 알 수 있다[6]. 하지만 많은 체외진단의료기기 제조사들이 개발 및 승인과 관련하여 임상적 성능시험과 인허가 과정을 가장 어려운 점으로 뽑았다[7]. 이에 본 연구에서는 체외진단기기의 임상적 성능시험뿐만 아니라 국내 식품의약품안전처 허가와 유럽 인증 과정의 실제 경험을공유하고자 하였다.
체외진단의료기기법이 2020년 5월 1일부터 시행되면서 국내 임상 검사실에도 많은 변화가 일어났다. 체외진단의료기기법에서는 임상적 성능시험을 체외진단의료기기의 성능을 증명하기 위하여 검체를 분석하여 임상적·생리적·병리학적 상태와 관련된 결과를 확인하는 시험으로 정의하고 있으며, 이러한 시험을 진행하기 위해서는 임상적 성능시험기관으로 지정받아야 하고 연구자들은 임상적 성능시험 종사자 교육 등을 연간 8시간씩 이수하여야 한다[1]. 식품의약품안전처의 허가를 받기 위해서는 임상적 성능시험을 포함한 여러 기술문서들을 제출하여야 하며 등급에 따라 일부 자료의 제출이 면제될 수는 있으나 대부분의 경우는 Table 3에 있는 항목들과 관련된 문서를 제출하여야 한다. 면제 가능한 항목은 체외진단의료기기 허가·신고·심사 등에 관한 규정 별표 7 (체외진단시약의 기술문서 등 제출 자료의 범위)과 8 (체외진단장비의 기술문서 등 제출 자료의 범위)에 안내되어 있다[2]. 또한 해당 제품 특성상 제출자료 일부가 불필요한 경우, 그 사유를 구체적으로 기재하면 자료 제출을 면제받을 수도 있다.
Table 3
List of technical documentation specified for each approval process
| 체외진단의료기기 허가 ·신고·심사 등에 관한 규정 제25조 제1항[2] |
|---|
| 1. 이미 허가 ·인증 받은 제품과 비교한 자료 |
| 2. 기원·개발경위, 검출 또는 측정원리·방법에 관한 자료 |
| 3. 국내·외 사용현황에 관한 자료 |
| 4. 원재료 및 제조방법에 관한 자료 |
| 5. 사용목적에 관한 자료 |
| 6. 저장방법과 사용기간 또는 유효기간에 관한 자료 |
| 7. 성능을 확인하기 위한 자료 |
| 가. 분석적 성능시험에 관한 자료 |
| 나. 임상적 성능시험에 관한 자료 |
| 다. 품질관리 시험에 관한 자료 |
| 라. 표준물질 및 검체보관 등에 관한 자료 |
| 8. 취급자 안전에 관한 자료 |
| In Vitro Diagnosis Medical Device Regulation (IVDR) ANNEX II and III [3] |
|---|
| ANNEX II |
| 1. DEVICE DESCRIPTION AND SPECIFICATION, INCLUDING VARIANTS AND ACCESSORIES |
| 1.1. Device description and specification |
| 1.2. Reference to previous and similar generations of the device |
| 2. INFORMATION TO BE SUPPLIED BY THE MANUFACTURER |
| 3. DESIGN AND MANUFACTURING INFORMATION |
| 3.1. Design information |
| 3.2. Manufacturing information |
| 4. GENERAL SAFETY AND PERFORMANCE REQUIREMENTS |
| 5. BENFIT-RISK ANALYSIS AND RISK MANAGEMENT |
| 6. PRODUCT VERIFICATION AND VALIDATION |
| 6.1. Information on analytical performance of the device |
| 6.2. Information on clinical performance and clinical evidence. Performance evaluation report |
| 6.3. Stability (excluding specimen stability) |
| 6.4. Software verification and validation |
| 6.5. Additional information required in specific cases |
| ANNEX III |
| 1. Post market surveillance plan |
| 2. Post market performance follow up plan |
| In Vitro Diagnosis Medical Device Directive (IVDD) ANNEX III [13] |
|---|
| - General description of the product |
| - Documentation of the quality system |
| - Design information |
| - Information on the origin of materials and on their conditions |
| - Descriptions and explanations needed to ensure understanding |
| - Results of the risk analysis |
| - Description of the procedures used in the case of sterile products |
| - Results of the design calculations and of the inspections carried out |
| - Essential requirements when combined with device(s) that have the characteristics specified by the manufacturer, in the case that the device is to be combined with other device(s) |
| - Test reports |
| - Adequate performance evaluation data |
| - Labels and instructions for use |
| - Results of stability studies |
![]()
본 연구에서 진행한 임상적 성능시험의 경우 높은 임상적 민감도와 특이도, 일치도를 보였으나, 임상적 성능시험에 사용된 검체를 양성과 음성으로만 구분하여 검체별 농도에 대한 평가는 진행하지 못하였다. 본 연구 당시에는 2020년 11월에 발행된 체외진단의료기기 임상적 성능시험 가이드라인을 참고하였고, 해당 가이드라인에는 다양한 농도를 포함한 임상검체를 이용하여 평가하는 것을 권고하고 있었다[11]. 그러나 2022년 7월에 새로 발행된 고위험성 감염체 체외진단시약 성능시험 가이드라인에서는 고농도 음성 및 저농도 양성 검체를 포함시키도록 하고 있어 체외진단의료기기의 임상적 성능시험도 점점 강화되고 있다는 것을 알 수 있다[12].
유럽에서는 1998년부터 체외진단의료기기지침이 시행되었고, 2022년 5월 26일부터 기존의 체외진단의료기기지침이 체외진단의료기기규정으로 강화되었다[3, 13]. 체외진단의료기기규정은 24개의 조(article)로 구성되어 있는데 반하여 체외진단의료기기규정은 10개의 장(chapter)과 113개의 조로 구성되어 있으며 14개의 부속서(annex)를 포함하고 있어 더 포괄적이고 상세한 규정을 포함하고 있다. 이뿐만 아니라 체외진단의료기기지침과 체외진단의료기기규정의 큰 차이는 체외진단의료기기의 등급분류와 그에 대한 심사 주체이다. 체외진단의료기기지침에서는 체외진단의료기기의 등급을 목록기반으로 구분하였으나, 체외진단의료기기규정은 국내와 비슷하게 위험도를 기반으로 등급을 구분하였다[14]. 또한 체외진단의료기기규정은 제조사가 아닌 인증기관으로 지정받은 제3의 기관이 기술문서 등을 심사를 하고 인증서(certificate)를 발급하도록 하고 있다[5, 15]. 위험도가 높아지는 class A (멸균제품)와 class B, C, D 등급의 승인을 신청하기 위해서는 임상적 성능을 입증할 수 있는 자료를 제출하여야 하며, 이때 요구하는 기술문서들 또한 체외진단의료기기지침과 비교하여 더 명확해지고 다양해졌다(Table 3) [16]. 임상적 증거(clinical evidence)의 수준을 구체적으로 설명하기 위하여 과학적 타당성(scientific validity)을 추가적으로 입증하도록 하였고, 임상적 성능시험의 기준도 강화되었다. 한편 임상적 성능을 입증하기 위한 자료로 임상적 성능시험 외에도 과학적 동료 검토 문헌(scientific peer reviewed literature)이나 진단적 검사를 통해 발표된 경험(published experience gained by routine diagnostic testing)도 인정하고 있으며, 유럽 인증 시에도 만약 해당 제품의 특성상 제출자료 일부가 불필요한 경우에는 그 사유를 구체적으로 기재하여 자료 제출을 면제받을 수 있다. 인증기관은 제출된 기술문서와 함께 품질경영시스템도 심사한다. 본 연구에서도 2022년 4월 1일 기술문서 제출 후 2022년 4월 19일부터 22일까지 4일간 인증기관이 제조사를 방문하여 현장 평가(on site assessment)를 진행하였다. 승인 후에도 class C와 D 등급 체외진단의료기기는 정기 안전 변경 보고서(periodic safety update report)를 정기적으로 제출하여야 한다. 이러한 위험관리(risk management)와 시판 후 감시 시스템(post market surveillance system) 등은 국내 식품의약품안전처 허가와도 차이를 보이며, 유럽의 경우 해외 배송의 안전성도 확인하기 위하여 선적 안정성 연구(shipping stability study) 등을 추가로 요구한다. 2023년 6월 23일 현재 체외진단의료기기 심사기관으로 지정되어 있는 인증기관은 모두 10개이다(Table 4) [17]. 심사기관이 충분하지 않다 보니 기존 체외진단의료지침하에 CE 마크를 획득했던 제품들이 다시 심사를 받고 승인되는데 지연이 되고 있으며 이로 인해 이행 유예기간을 등급에 따라 최대 2028년 5월 26일까지 연장하기도 하였다[18]. 또한 이번 연구에 사용된 시약같이 제조사의 등록 영업소가 유럽 회원국 내에 있지 않은 경우에는 반드시 유럽대리인을 지정해야 한다. 유럽대리인은 자가신고와 관련된 서류를 검증하여야 하고, 자가선언 및 승인이 완료된 제품의 지속적인 관리의 책임도 갖게 된다. 본 연구과정에서 승인된 NextGene MTB/NTM Detection Kit는 새로운 체외진단의료기기규정하에서 받은 국내 최초의 class C 등급 인증으로 앞으로 많은 제조사와 기관들이 유럽 인증을 진행하는데 본 연구가 도움이 될 수 있길 희망한다.
Table 4
Lists of notified bodies under regulation (EU) 2017/746 on in vitro diagnostic medical devices
![]()
2022년 5월 26일 이후 체외진단의료기기지침에 따른 자가선언이나 인증은 금지되었다. NextGene STI 12 Detection Kit 3&4는 그 이전인 2022년 4월 7일에 자가선언을 통해 등록되었다. 따라서 유예기간이 끝나는 2026년 5월 26일 이전까지 체외진단의료기기규정에 따라 class C로 인증을 변경하여야 한다. 체외진단의료기기지침에서도 4등급(others, self testing, List B, Lits A)으로 분류하였으며, NextGene STI 12 Detection Kit 3&4는 자가선언을 통해 others 등급으로 등록되어 있다. 체외진단의료기기지침은 List B와 List A 등급만 인증기관에 의한 심사와 인증을 요구하였으나, 새로 시행된 체외진단의료기기규정은 일부 class A를 제외한 class B, class C, class D 등급에서 심사와 인증을 요구하고 있다. 이로 인해 체외진단의료기기지침하에서는 10–20%의 제품만이 심사를 받고 나머지 제품들은 자가선언을 통해 등록할 수 있었던 것에 반해 체외진단의료기기규정하에서는 80–90%의 제품이 심사를 통해 인증이 이루어지게 되어, 보다 엄격하게 체외진단의료기기를 관리하게 되었다[19, 20]. 때문에 기존 체외진단의료지침하에 CE 마크를 획득한 제품이라도 다시 CE 마크를 받기 위해서는 임상적 성능시험과 같은 더 많은 준비가 필요하게 되었다.
임상적 성능시험은 새로 개발된 체외진단의료기기를 평가하는데 중요한 과정이며, 국내 식품의약품안전처 허가와 유럽 인증 과정 모두에서 요구하는 절차 중에 하나이다. 하지만 앞서 언급한 대로 많은 기업체들은 국내 혹은 해외 임상적 시험평가에 어려움을 겪고 있으며, 특히 임상적 성능시험을 진행하는데 있어서는 임상적 성능시험기관과의 매칭을 가장 큰 애로사항으로 뽑았다[7]. 이와 관련하여 한국보건산업진흥연구원에서는 체외진단의료기기 신속 임상시험 지원 서비스를 통해 기업과 필요한 검체 매칭을 지원하고 있기도 하다. 그러나 검체를 수집하는데 많은 시간이 필요하고 임상적 성능시험을 진행하기 위해서는 많은 비용이 들어 중소기업에서 제품을 개발하는데 여전히 어려움이 크다. 정부에서 직접 인체유래물은행 등을 설립 및 운영한다면, 좀 더 원활하고 신속한 임상적 성능시험이 이루어질 수 있을 것이다[21]. 2019년 이후 국내에서는 코로나19의 확산으로 인한 철저한 개인위생이 이루어지면서 인플루엔자 바이러스의 감염이 급격하게 줄었다[22]. 때문에 인플루엔자 바이러스 확진자의 검체를 구하는 것이 매우 힘들어졌으며 2019년 이전에 보관해 놓았던 잔여 검체를 새로운 임상적 성능시험에 사용하기 위해서는 확진 당시 사용하였던 동일한 장비 및 시약을 사용하여 보관되어온 검체가 확진 당시와 동등한 Ct (cycle threshold) 값을 갖는다는 것을 증명해야 하는 등의 어려움이 있다. 또한 국내에 부족한 임상 검체의 경우 피험자 수를 완화하거나 해외에서 수급할 수 있도록 지원해 주는 시스템의 도입이 필요하다[7]. 경쟁력 있고 원활한 제품 개발 및 인허가를 위해서는 제조사, 식품의약품안전처, 진단검사의학회 등이 함께 고민하는 것이 필요하다.
국내 체외진단의료기기법은 등급에 따라 신고, 인증, 허가로 처리 절차를 구분하고 있으며 식품의약품안전처에서 주관하고 있다. 한국법제연구원 산하 법령번역센터에서 제공하는 영문 체외진단의료기기법에서는 허가를 permission으로 번역하고 있으나 일부 식품의약품안저처에서 발표한 영문 공식 성명서에서는 허가를 approval로 번역하고 있다[1, 23]. 본 논문에서는 일반적으로 권한을 부여한다는 의미를 지닌 permission보다는 approval이 허가의 의미를 더 잘 전달할 수 있다고 여겨 허가를 approval로 번역하였다. 또한 본 논문에서는 국내에서의 허가와 유럽에서의 인증으로 용어를 구별하였다. 국내의 경우 상대적으로 위해성이 낮은 1, 2등급의 경우 임상적 성능시험 자료를 요구하지 않으며 식품의약품안전처에서 의료기기안전정보원으로 업무를 위임하여 처리하고 있다. 1등급은 신고, 2등급은 인증을 통해 처리된다. 반면 위해성이 있는 2등급 및 3, 4등급의 경우 식품의약품안전처에서 심사를 거쳐 허가하고 있으며, 이때 임상적 성능시험 자료가 함께 제출되고 심사되어져야 한다. 유럽의 경우는 자가선언 혹은 인증으로 절차를 구분하며 인증의 경우 인증기관의 심사를 거쳐 인증서를 받게 되며, 이렇게 인증된 제품만이 CE 마크를 부착할 수 있다. 이렇게 국내 체외진단의료기기법과 유럽 체외진단의료기기규정에서 사용하는 인증의 의미가 다르기 때문에 주의가 필요하다.
코로나19 팬데믹 시기를 겪는 동안 많은 임상 검사실들은 분자진단검사실을 추가하였으며, 실제로 2020년 5월 13일까지 총 229개 기관이 코로나19 진단검사 인증을 받았다[24]. 또한 진단검사의학재단 우수검사실 분자진단검사 인증을 신청한 기관이 2019년에는 151기관이었으나 2022년에는 267기관으로 증가하여 다른 분야에 비해 훨씬 높은 증가율을 보였다(Supplementary Table 1). 이는 많은 검사실들이 검사장비, 음압실 등 분자진단검사를 진행하기 위한 시설을 새로 갖추었음을 의미하며, 검사를 수행할 수 있는 숙련된 임상병리사가 확보되었다는 의미이기도 하다. 대한진단검사의학회 또한 유관학회, 산업계, 정부 등과 함께 유기적인 협력을 바탕으로 코로나19의 분자진단검사법을 조기에 정착시키는데 크게 이바지하였을 뿐 아니라 질병관리청 등과 함께 코로나19 진단을 위한 여러 가이드라인을 제시하고 지속적으로 업데이트하였다[24-27]. 이러한 인적·물적 자원과 경험을 잘 활용한다면 진단 검사법의 발전과 급성장하고 있는 체외진단의료기기 산업에 많은 도움이 될 것이다.
본 연구는 임상적 성능시험을 포함한 전체 국내 및 유럽 허가(인증) 과정을 포함하고 있다. 앞서 언급한 바와 같이 임상 연구자, 산업계, 정부 등이 유기적인 협력을 통해 어려운 점들을 개선해 나가고, 본 연구와 같이 실제 허가(인증) 과정에 대한 경험이 쌓이면 국내 체외진단의료기기 산업은 더욱더 발전하고 경쟁력 있는 제품을 개발할 수 있을 것이다.
 XML Download
XML Download