

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Hemophagocytic lymphohistiocytosis (HLH) is a rare life-threatening hematological disorder characterized by uncontrolled activation of CD8+ T and natural killer cells, cytokine storm, and uncontrolled hemophagocytosis, leading to severe organ dysfunction [1]. It can be classified into two groups: primary and secondary. Primary HLH is caused by genetic mutations in cell-mediated cytotoxicity, while the latter is a clinical syndrome associated with infection, malignancy, drugs, and rheumatic diseases. Secondary HLH associated with autoimmune and autoinflammatory diseases is called macrophage activation syndrome (MAS), which shows a good response to immunosuppressive therapy for the underlying conditions. However, there is no consensus regarding the treatment of MAS that is unresponsive to conventional immunosuppressive treatments [2].
Treatment of secondary HLH is complicated and controversial. While the standard care for primary HLH is induction therapy per the “HLH-2004 protocol,” including dexamethasone, etoposide, cyclosporine, and intrathecal methotrexate, the approach has not been fully validated in patients with secondary HLH [3]. Because there is no established treatment of choice, there have been several reports of attempts involving the use of other immunosuppressants such as interleukin (IL)-1 inhibitors, IL-6 inhibitors, and anti-interferon-γ (anti-IFN-γ) agents [4]. Another potentially interesting approach is the use of Janus kinase (JAK) inhibitors [5]. After the binding of various cytokines to their receptors, JAKs activate signal transducers and activators of transcription (STATs), which dimerize and enter the nucleus and stimulate the expression of genes related to cell survival, differentiation, and proliferation. The cellular targets of JAK inhibitors include various components of both the innate and adaptive immune systems, such as natural killer cells, dendritic cells, and T cells [6]. Recently, a single-center pilot trial showed that ruxolitinib, an oral JAK1 and JAK2 inhibitor, effectively controlled secondary HLH. However, the efficacy of ruxolitinib has not yet been thoroughly evaluated in patients with refractory MAS.
Herein, we describe a case of HLH refractory to glucocorticoids (GCs) and cyclosporine that was successfully treated with ruxolitinib in a patient with systemic lupus erythematosus (SLE).
CASE REPORT
A 64-year-old woman with no specific underlying disease was hospitalized in the rheumatology department with complaints of general weakness and fever that started 6 months ago. She also complained of polyarthralgia, poor oral intake, and loss of body weight (–10 kg) during the previous 6 months. Physical examination revealed a body temperature of 38.7°C and swelling and tenderness of the proximal interphalangeal and radiocarpal joints. Laboratory examination revealed leukopenia (1.33×103/µL with an absolute neutrophil count of 904/µL) and an anemia (hemoglobin level: 8.9 g/dL) with a positive Coombs test. The patient tested positive for antinuclear antibodies, with a titer of 1:320. Serum complement (C3 and C4) levels were markedly low (48 and 3 mg/dL, respectively), and the anti-double-stranded DNA titer was 2,450 IU/mL. Urine analysis revealed proteinuria, with a urine protein-to-creatinine ratio of 1.76. Contrast-enhanced computed tomography revealed multiple neck and mediastinal lymphadenopathies, suggesting reactive changes. Additionally, hepatomegaly was significantly present, while splenomegaly was not prominent. Bone marrow examination conducted to further evaluate the patient's leukopenia revealed an increased number of polyclonal plasma cell on bone marrow section. However, bone marrow aspirate was diluted with peripheral blood, which was not appropriate for precise evaluation. Because lupus nephritis was suspected, kidney biopsy was performed on the third day of hospitalization. Renal histology revealed focal proliferative glomerulonephritis. Based on these findings, the patient was diagnosed as having SLE with kidney involvement and treated with GC pulse therapy (500 mg/day of methylprednisolone for 3 days) and mycophenolate mofetil. Following pulse therapy, the patient continued high-dose GC treatment, and her fever and arthralgia resolved.
However, on the fifth day of hospitalization, she developed high fever (up to 39.0°C) and hypotension and lost consciousness. Laboratory findings at that time are presented in Table 1. Briefly, serum levels of ferritin, soluble IL-2 receptor, aspartate transaminase (AST), and alanine transferase (ALT) were 15,767 mg/dL (reference range: 4.6~204.7 ng/mL), 4,831 IU/mL (reference range: 158~623 IU/mL), 69 IU/mL, and 101 IU/mL, respectively. No bacterial growth was observed in blood or urine cultures. Based on the clinical (fever) and laboratory findings (bicytopenia at hospital day 1, high level of ferritin, triglyceride, and soluble IL-2 receptor), a diagnosis of HLH was made according to the HLH-2004 diagnostic criteria [3,7,8]. The hemophagocytic syndrome score (HS-score) at this time was 224, which indicates that the probability of HLH is between 96% and 98% [9].
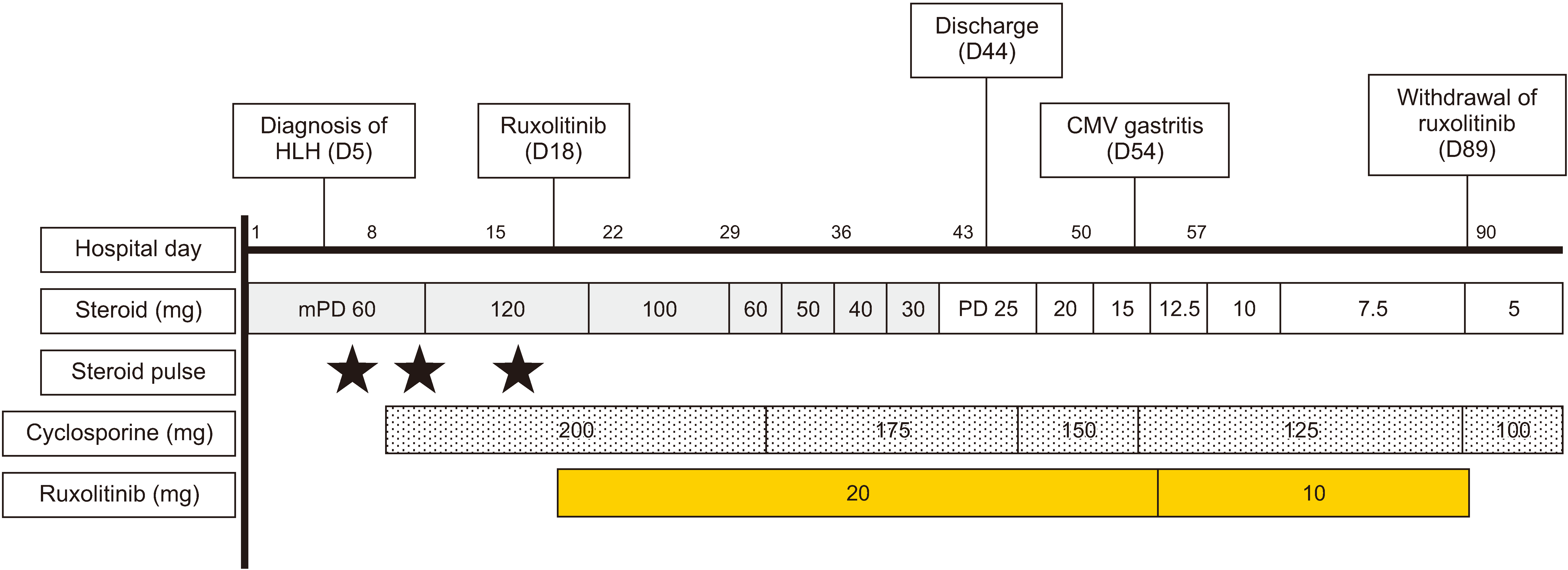
The patient received another course of GC pulse therapy and cyclosporine (100 mg, twice daily). After treatment, her level of consciousness transiently improved; however, she became drowsy again 2 days after the completion of the pulse therapy. Serum ferritin levels peaked at 239,325 mg/dL, and AST/ALT levels continuously increased. Based on the above clinical features suggestive of refractoriness to GC and cyclosporine, the patient was treated with ruxolitinib (10 mg twice daily) starting from hospital day 18. After treatment, her symptoms, level of consciousness, and hypotension markedly improved and her ferritin level gradually decreased to 25,325 g/dL on hospital day 20 (Figures 1 and 2). Asymptomatic antigenemia (17 infected cells per 200,000 white blood cell) caused by cytomegalovirus (CMV) infection was detected on hospital day 40. Fundoscopic examination and sigmoidoscopic evaluation revealed no evidence of CMV infection. Preemptive ganciclovir treatment was considered; however, the patient wanted to be discharged and receive antiviral treatment if the antigenemia titer increased in subsequent testing. Based on a shared decision with the patient, she was discharged on hospital day 44 and scheduled to visit the clinic 1 week later.
One week after discharge, the patient visited the clinic complaining of nausea and anorexia. She was hospitalized again, and endoscopic examination of the stomach revealed shallow ulcerations on the distal lesser curvature. Pathological features of the gastric biopsy specimen were consistent with those of CMV gastritis. The patient was treated with ganciclovir, and ruxolitinib was tapered to a dose of 10 mg once daily. There was no HLH flare or underlying SLE after tapering the ruxolitinib dose. The patient’s gastrointestinal symptoms improved shortly after ganciclovir treatment started, and she received the treatment for 5 weeks. During the follow-up, other adverse events related to ruxolitinib did not occur.
Three months after the first hospitalization, ruxolitinib was discontinued because of persistent disease remission. The doses of cyclosporine and GC were gradually tapered, and the patient’s underlying nephritis was in remission. Currently, 3 years after the diagnosis of HLH, the patient is being followed up at a rheumatology clinic with low-dose GC and cyclosporine treatment.
DISCUSSION
Although secondary HLH is associated with poor prognosis and high mortality rate, the optimal treatment option has not been thoroughly investigated [10-12]. Recently, there have been a few reports of favorable treatment outcomes with rituximab, anakinra, and IV immunoglobulin in patients with refractory HLH, although most were case reports.
HLH is often associated with a dysregulated immune response that results in the activation and proliferation of T cells and macrophages, leading to a cytokine storm and systemic inflammation. Because the overproduction of T cells mediates IFN-γ activity and the activation of JAK-STAT is a critical pathway in the pathogenesis of HLH, blocking the pathway can be a potential therapeutic target. It also suggests that targeting a single cytokine or cellular component could be inadequate to control the disease. Of note, a few recent prospective controlled studies have investigated the use of ruxolitinib in patients with secondary HLH and reported promising results [13]. In an open-label pilot trial, five adult patients with secondary HLH received ruxolitinib and were followed for a median period of 490 days. There were no cases of death during the observation and all patients achieved partial or complete remission with a favorable safety profile [5]. Based on this result, we selected ruxolitinib for the treatment of refractory HLH in this case. To the best of our knowledge, this is the first report of ruxolitinib investigation in a patient with HLH secondary to SLE in South Korea. The patient’s neurological manifestations, fever, and laboratory abnormalities rapidly improved, and the GC dose was successfully tapered. Notably, proteinuria and urinary casts disappeared after ruxolitinib treatment. This suggests that blocking of the JAK-STAT pathway could be a potential therapeutic strategy for treating SLE.
Although ruxolitinib treatment markedly improved SLE and secondary HLH, CMV gastritis developed 8 weeks after treatment. Because the inhibition of the JAK-STAT pathway significantly interferes with T-cell function, ruxolitinib could reduce the host response to viral pathogens and increase the risk of infection [14]. Previous studies investigating the efficacy of ruxolitinib in patients with steroid-refractory graft-versus-host disease showed that CMV reactivation occurs in 10%~30% of patients [7,15]. Additionally, patients with HLH receiving ruxolitinib are concomitantly administered other immunosuppressive treatments, which may further increase the risk of CMV disease. Therefore, in patients with secondary HLH receiving ruxolitinib, careful monitoring for CMV reactivation should be performed to improve treatment outcomes.
SUMMARY
In summary, we reported a case of refractory HLH secondary to SLE that was successfully treated with ruxolitinib. Although the efficacy and safety of ruxolitinib for the treatment of SLE-related HLH should be further investigated in future studies, our case suggests that it may be a promising treatment option.
 XML Download
XML Download