INTRODUCTION
In oncology, molecular profiling of tumors provides vital information for diagnosis and treatment. Among various somatic molecular aberrations, structural variants (SVs) in chromosomes play important roles in tumor development and progression. Notably, in hematologic malignancies, a primary mechanism of oncogenesis is the formation of fusion genes by chromosomal rearrangements, resulting in loss of control over cell division and proliferation [
1]. Therefore, SVs are important markers for diagnosis, therapy selection, and predicting prognosis through risk stratification in hematologic malignancies [
2].
SVs are largely composed of unbalanced copy number variants (CNVs) (i.e., insertions, duplications, and deletions) and balanced rearrangements (i.e., inversions and translocations), with sizes ranging from 50 bp to over megabase pairs [
3]. Conventional tests, including chromosomal banding analysis (CBA), FISH, chromosomal microarray (CMA), reverse transcription PCR (RT-PCR), and multiplex ligation-dependent probe amplification (MLPA), are performed in combination to detect these SVs. However, these tests have limitations. CBA has a low resolution, requires fresh samples, and involves a 7–10-day processing time because of the mandatory cell culturing process. For FISH and RT-PCR, probes and primers target known variants and cannot detect novel variants or rare breakpoints. These methods are also labor-intensive and lack multiplexing capacity. Despite its high resolution and capability to test the whole genome, CMA cannot detect balanced chromosomal aberrations and determine where an insertion has occurred when copy numbers increase.
Next-generation sequencing (NGS) has recently been adopted to identify genetic variants, including SVs, in hematologic malignancies [
4-
6]. However, its common short-read platform, with a read length of 150–300 bp, has limitations in precisely analyzing large SVs and homologous elements such as repetitive sequences and pseudogenes [
7].
There is growing interest in optical genome mapping (OGM), a single-molecule strategy that can analyze tens to hundreds of kilobase-long reads, as a potential alternative technology for analyzing SVs [
3,
8]. In OGM, fluorescent markers are used to tag particular sequences within DNA fragments of up to 1 megabase in length. This technique enables
de novo assembly and gap filling and can detect SVs of up to tens of kilobases long [
9-
11]. Differences in the fluorescent labeling patterns relative to a consensus genome map are used to identify SVs [
12]. OGM has the potential to overcome the limitations of conventional methods and detect SVs with a higher resolution in a substantially shorter time. Moreover, OGM can help identify novel chromosomal aberrations that may be used as additional markers for diagnosis, targeted therapy, and prognosis prediction.
We analyzed OGM results of diagnostic samples from patients with hematologic malignancies and evaluated their concordance with the results of conventional methods in detecting SVs. Several recent studies have assessed the clinical usefulness of SV detection using OGM in patients with blood cancer [
13-
21]. These studies mainly used CBA, FISH, RT-PCR, CMA, or MLPA as conventional methods for comparison. However, studies using RNA sequencing as a method for comparison, like our study, are scarce [
20]. In particular, our study is the first report to assess the feasibility of OGM for SV detection in hematologic malignancies in Korea.
RESULTS
Samples
In total, 27 BMA samples of hematologic malignancies, including AML (N=9), acute promyelocytic leukemia (APL; N=1), CML in the chronic phase (CML-CP; N=3), primary myelofibrosis (PMF; N=1), MDS with excess blasts-2 (MDS-EB-2; N=1), MDS with single lineage dysplasia (MDS-SLD; N=1), B-lymphoblastic leukemia (B-ALL; N=6), CLL (N=1), marginal zone B-cell lymphoma (MZBCL; N=1), lymphoplasmacytic lymphoma (LPL; N=1), and plasma cell myeloma (PCM) (N=2), were analyzed. The median age of the study cohort was 55 yrs (range: 3–84 yrs); 13 samples were from men, and 14 were from women. The 27 samples were analyzed using conventional methods, including karyotyping (27/27), FISH (7/27), RT-PCR (11/27), and RNA fusion panel analysis (14/27).
Samples were classified into simple or complex cases based on the number of SVs detected using the conventional methods (simple: 0–2; complex: ≥3). Of the 27 cases, 18 were classified as simple and nine as complex. In total, 68 SVs (18 aneuploidies, 28 translocations, 16 deletions, two duplications, one inversion, one insertion, one marker chromosome, and one isochromosome) were detected.
OGM data quality
OGM of the 27 samples resulted in an average DNA amount of 1,220.8 Gbp, an average 234.65-fold effective coverage, an average N50 of 0.208 Mbp, an average label density of 15.24/100 kb, and an average mapping rate of 60.2%. For
de novo assembly for SV analysis, the minimum recommended coverage for detecting heterozygous and homozygous SVs is 80×. Twenty-six of the 27 samples met this recommendation, and the results of all samples were compared with those from the conventional methods. The data quality of all cases is provided in
Supplemental Data Table S2. No SVs were detected by OGM in the four negative controls.
OGM in simple cases
In the 18 simple cases (seven AML, one APL, three CML-CP, one PMF, five B-ALL, and one CLL), the conventional methods detected no SVs in one case, one in 14 cases, and two in three cases (
Table 1), totaling 20 SVs. OGM detected 17 (85%) SVs correctly, only part of the aberration in the three remaining (15%) SVs, and 10 additional SVs. Overall, OGM showed concordance in 15 (83.3%) cases and partial concordance in three (16.7%) cases. In the 15 concordant cases, in which OGM could detect all variants identified using conventional methods, five cases also had additional SVs detected uniquely by OGM.
Table 1
Comparison of routine diagnostic test results with OGM results in simple cases
|
Sample ID |
Diagnosis |
Karyotype |
FISH |
RT-PCR/RNA fusion |
OGM results
(SV tool and/or CNV tool) |
Summary |
|
2 |
AML |
46,XX[20] |
NA |
NUP98::NSD1
|
t(5;11)(q35.3;p15.4) NUP98-NSD1
|
Concordant |
|
3 |
AML |
46,XY,inv(16)(p13q22)[23]/46,XY[2] |
NA |
CBFB::MYH11
|
t(16;16)(p13.11;q22.1) CBFB-MYH11
|
Concordant |
|
4 |
AML |
46,XX,der(9)ins(9;8)(q13;q22q24.2)[22]/46,XX[1] |
NA |
NA |
MYC amplification |
Concordant
|
|
5 |
AML |
47,XX,+8,t(9;11)(p21;q23)[20] |
NA |
KMT2A::MLLT3
|
+8, t(9;11)(p21.3;q23.3) KMT2A-MLLT3
|
Concordant |
|
6 |
AML |
46,XX,t(11;12)(p15;q13)[20] |
NA |
NUP98::HOXC13
|
t(11;12)(p15.4;q13.13) NUP98-HOXC13
|
Concordant |
|
8 |
AML |
46,XY,dup(19)(p13.1p13.3)[20]/46,XY[6] |
NA |
Negative |
dup(19)(p13.2p13.2) |
Concordant |
|
9 |
AML |
46,XX,t(6;9)(p22;q34)[16]/46,XX[4] |
NA |
DEK::NUP214
|
t(6;9)(p22.3;q34.13) DEK-NUP214
|
Concordant |
|
10 |
APL |
46,XY,t(15;17)(q24;q21)[19]/46,XY[1] |
NA |
PML::RARA
|
t(15;17)(q24.1;q21) PML-RARA
|
Concordant |
|
11 |
CML-CP |
46,XY,t(9;22;11)(q34;q11.2;q13)[20] |
NA |
BCR::ABL1(b3a2, major type) |
t(9;22)(q34.12;q11.23) BCR-ABL1
t(11;22)(q13.1;q11.23) ARL2-SNX15-CABIN1
t(9;11)(q34.11;q13.1)
|
Concordant
Additional by OGM
|
|
12 |
CML- CP |
46,XX,t(3;11;9;22)(p21;q13;q34;q11.2)[20] |
NA |
BCR::ABL1(b2a2, major type) |
t(9;11)(q34.11;q13.2)
t(9;22)(q34.12;q11.23)
t(3;22)(p21.31;q11.23)
|
Partial concordance |
|
13 |
CML-CP |
46,XY,t(7;9;22)(q22;q34;q11.2)[14] |
NA |
BCR::ABL1(b2a2, major type)
BCR::ABL1(e1a2, minor type,weakly positive)
|
t(7;22)(q22.1;q11.23)
t(9;22)(q34.12;q11.23)
|
Partial concordance |
|
14 |
PMF |
46,XY,der(6)t(1;6)(q21;p21)[3]/46,XY[17] |
NA |
NA |
del(6)(p21.1p25.3) |
Partial concordance |
|
17 |
B-ALL |
46,XY,t(9;22)(q34;q11.2)[3]/47,idem,+21[9]/46,XY[8] |
NA |
BCR::ABL1(e1a2, minor type) |
t(9;22)(q34.12;q11.23) BCR-ABL1+21IKZF1 exon 4-7 deletion†SETD2 exon 1-10 deletion |
Concordant
|
|
18 |
B-ALL |
46,XY[30] |
Negative for t(11q23) break-apart |
Negative§
|
del(12)(p13.31p12.1)
del(17)(p11.2p13.3)
dup(17)(p11.2q25.3)
t(12;22)(p13.31;q13.2) EP300-ZNF384
|
Concordant
|
|
19 |
B-ALL |
47,XX,+X,t(2;14)(p11.2;q32)[5]/46,XX[20] |
NA |
Negative for BCR::ABL1
|
+X, t(2;14)(p11.2;q32.33) |
Concordant |
|
21 |
B-ALL |
46,XY,t(2;12)(p11.2;p13)[5]/46,XY[4] |
NA |
Negative for BCR::ABL1
|
t(2;12)(p12;p13.2)IKZF1 exon 4-7 deletion†CDKN2A whole gene deletionCDKN2B whole gene deletion |
Concordant
|
|
22 |
B-ALL |
46,XY[13] |
NA |
ETV6::RUNX1
|
t(12;21)(p13.2;q22.12) |
Concordant |
|
23 |
CLL |
46,XY[20] |
Positive for del(13q) |
NA |
del(13)(q14.3) |
Concordant |

The three simple cases that showed partial concordance were S12, S13, and S14. Cases S12 and S13 were CML-CP cases with complex translocations, harboring four-break and three-break translocations, respectively. In S12, t(3;11;9;22)(p21;q13;q34;q11.2) was reported by CBA but missed by OGM [t(3;11)(p21;q13)]. Similarly, t(7;9;22)(q22;q34;q11.2) was reported by CBA in S13, but t(7;9)(q22;q34) was missed by OGM. For S14, a case of PMF, der(6)t(1;6)(q21;p21) was reported by CBA, whereas OGM missed t(1;6)(q21;p21) and 1q21q44 gain.
Additional findings by OGM in simple cases
OGM revealed additional findings in five simple cases: S4, S11, S17, S18, and S21. S4 was an AML case; S11 was a CML-CP case; and S17, S18, and S21 were B-ALL cases. The karyotyping result of S4 was initially reported as 46,XX,ins(9)(q13p13p24)[
22]/46,XX[
1] or 46,XX,ins(9)(q13q21q13)[
22]/46,XX[
1]. OGM revealed a
MYC amplification as the inserted component, which was confirmed using FISH and NGS CNV analyses (
Fig. 1). Based on this, we modified the karyotyping result as 46,XX, der(9)ins(9;8)(q13;q22q24.2)[
22]/46,XX[
1]. In S11, in addition to the three translocations associated with the three-break balanced translocation and the
BCR::ABL1 fusion detected using conventional methods, OGM identified the
ARL2-SNX15::CABIN1 fusion. In S17, OGM correctly detected t(9;22)(q34;q11.2)
BCR::ABL1 fusion and trisomy 21 identified using the conventional methods and further detected
SETD2 exon 1–10 deletion. NGS CNV analysis results were reviewed to confirm the new finding, and
SETD2 exon 2–9 deletion and
IKZF1 exon 4–7 deletion were detected. The
IKZF1 exon 4–7 deletion, which was not called by OGM and was likely missing from the OGM results, was detected upon manual inspection of the OGM profile. For S18, CBA revealed a normal karyotype, and a false-positive call for the
ATP5L::KMT2A fusion was reported by the RNA fusion panel. The
ATP5L::KMT2A fusion was confirmed as a false positive by RT-PCR. OGM results confirmed the false-positive call of
ATP5L::KMT2A and found additional variants of del(12)(p13.31p12.1), del(17)(p11.2p13.3), dup(17)(p11.2q25.3), and
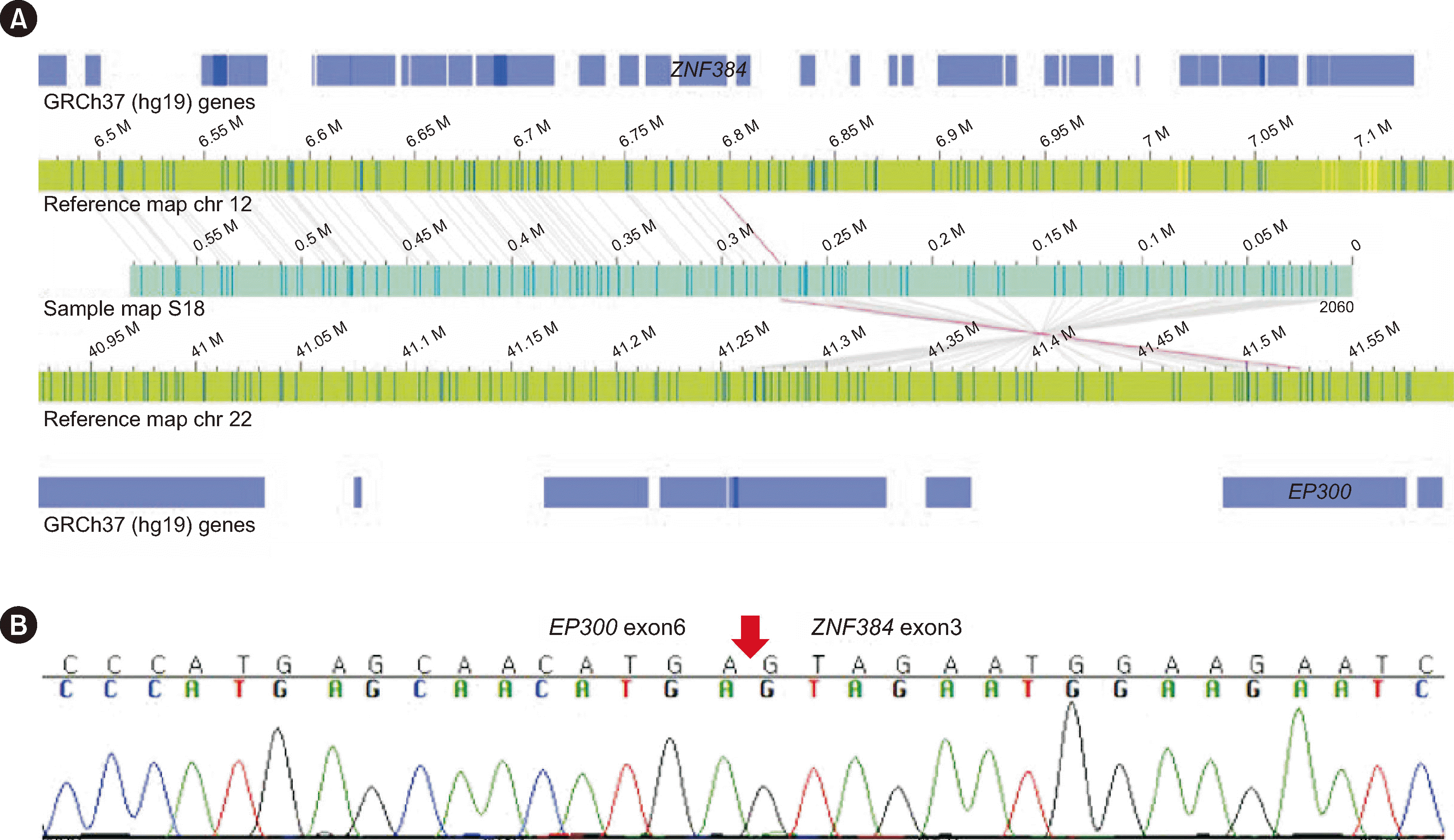
EP300::ZNF384 fusion.
EP300::ZNF384 was confirmed using Sanger sequencing, and del(12)(p13.31p12.1), del(17)(p11.2p13.3), and dup(17)(p11.2q25.3) were confirmed using NGS CNV analysis (
Fig. 2). In S21, OGM additionally identified
CDKN2A and
CDKN2B whole gene deletions, and NGS CNV results confirmed
CDKN2A whole gene deletion,
CDKN2B exon 2 deletion, and
IKZF1 exon 4–7 deletion. Notably, the deletion of
IKZF1 exon 4–7, which was not called by OGM, was detectable upon manual inspection of the OGM profile, as in the case of S17.
Fig. 1
Results of conventional laboratory tests and OGM for S4. (A) CBA revealed an insertion in the long arm of chromosome 9. (B) Whole-genome circos plot obtained by OGM showing MYC amplification. (C) FISH was performed using Vysis probes (Abbott Molecular). (D) NGS CNV analysis showing amplification of the q24.21 band in chromosome 8.
Abbreviations: OGM, optical genome mapping; CBA, chromosomal banding analysis; chr, chromosome; CNV, copy number variant.
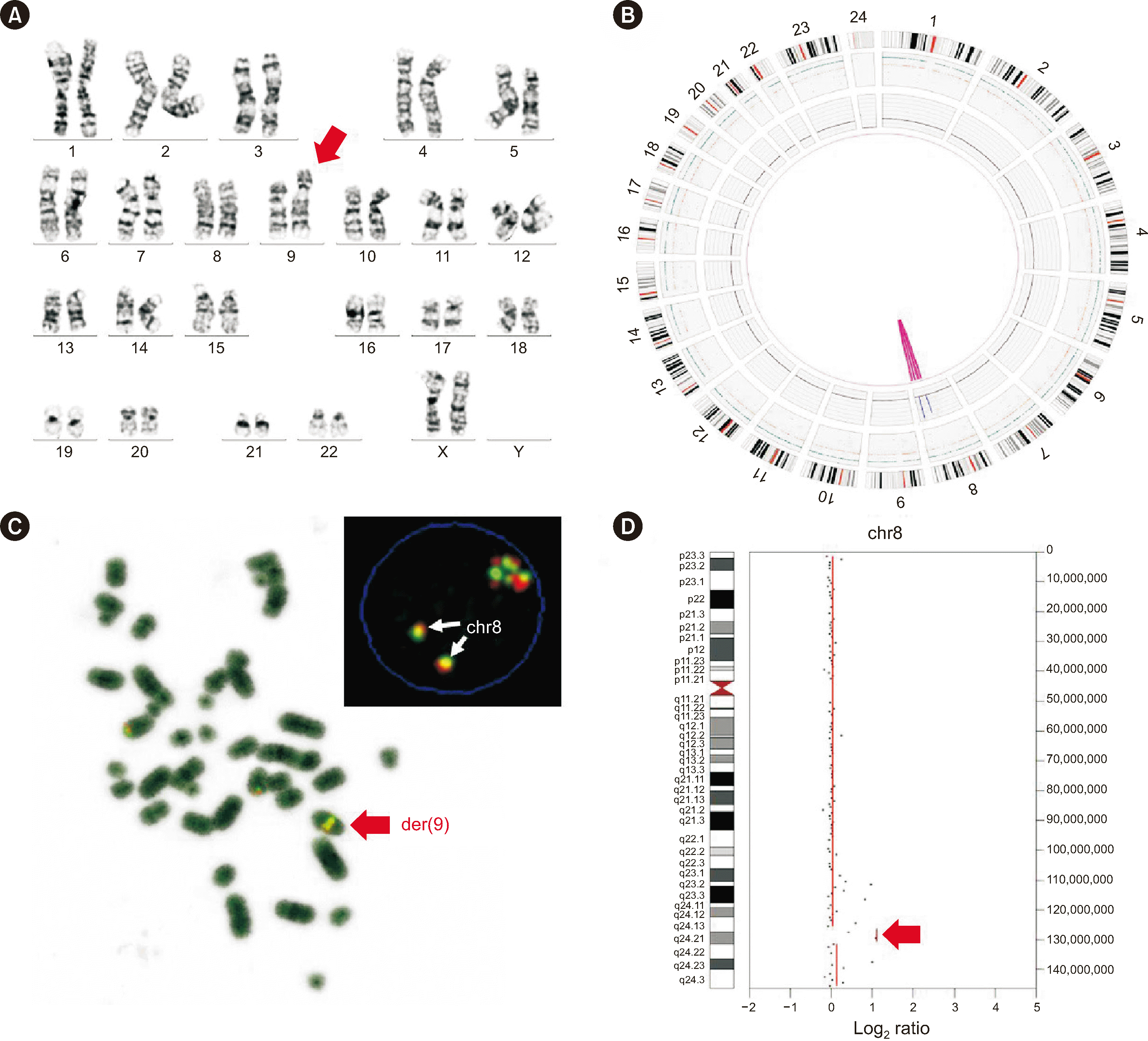

Fig. 2
Additional finding of EP300::ZNF384 fusion by OGM in S18. (A) Genome map view showing the EP300::ZNF384 fusion. Map of S18 (blue) aligned to reference maps of chromosome 12 and chromosome 22 (green), with breakpoints in ZNF384 and EP300, respectively. Label alignments between the reference and sample genome maps are indicated by gray lines. Translocation breakpoints are indicated by pink lines. (B) Confirmation of the EP300::ZNF384 fusion using Sanger sequencing. The red arrow indicates the breakpoint.
Abbreviations: OGM, optical genome mapping; chr, chromosome.
OGM in complex cases
In the nine complex cases (two AML, one MDS-EB-2, one MDS-SLD, one B-ALL, one MZBCL, one LPL, and two PCM), conventional methods detected three to 11 SVs in each case, totaling 48 SVs (
Table 2). OGM accurately detected 35 (72.9%) SVs, partially identified four SVs (8.3%), and failed to detect nine (18.8%) SVs. Overall, OGM demonstrated concordance in two (22%) cases and partial concordance in seven (78%).
Table 2
Comparison of routine diagnostic test results with OGM results in complex cases
|
Sample ID |
Diagnosis |
Karyotype |
FISH |
RT-PCR/RNA fusion |
OGM results
(SV tool and/or CNV tool) |
Summary |
|
1 |
AML |
46,XY,del(5)(q13q31),del(7)(q11.2),+11,der(12)t(1;12)(p13;q24.3),?psu dic(17;16)(p11.2;q21),?der(18)t(18;19)(q23;q13.1-13.3)[20] |
NA |
Negative |
del(5)(q14.3q34), del(7)(q11.23), +11, t(1;12)(p13.3;q24.33), t(17;16)(p11.2;q21), t(18;19)(q23;q13.2) |
Concordant |
|
7 |
AML |
45,XX,del(11)(q14q24),-16,der(21)t(16;21)(p11;q22)[20] |
NA |
FUS-ERG
|
del(11)(q14.1q24.1), -16, t(16;21)(p11.2;q22.2) FUS-ERG
|
Concordant |
|
15 |
MDS-EB-2 |
48,XY,+8,+der(10)t(1;10)(q21;q11.2)[4]/49,sl,+9[14]/50,sdl1,+6[4]/46,XY[2] |
Positive for trisomy 8 |
Negative |
+8, dup(1)(q21.1q44), dup(10)(p11.1p15.3), dup(10)(q11.2) +9, +6 |
Partial concordance |
|
16 |
MDS-SLD |
45,XX,der(2)t(2;14)(q35;q13),del(5)(q14),del(11)(q14),del(13)(q13q22),-14[3]/46,XX[16] |
NA |
NA |
del(2)(q35q37.3), del(5)(q31.1), del(11)(q12.2) |
Partial concordance |
|
20 |
B-ALL |
48-52,XX,dup(1)(q32q21),+4,+6,+9,+der(14)t(14;21)(q21.1;q21.2),+21,+21[cp6]/46,XX[29] |
NA |
P2RY8-CRLF2
Negative for BCR-ABL1
|
dup(1)(q31.3q21.2), +4, +6, +9, +21, +21, t(14;21)(q21.1;q21.2), dup(14)(q11.2q21.1), dup(21)(q21.2q22.3) |
Partial concordance |
|
24 |
MZBCL |
46,X,-X,t(3;7)(q27;q32),?+der(6;9)(p10;p10), -17,+mar[11]/46,XX,t(3;7)[4]/46,XX[3] |
NA |
NA |
t(3;7)(q27.2;q32.2) |
Partial concordance |
|
25 |
LPL |
47,XX,+i(5)(p10),del(7)(q32)[18]/47,idem,del(16)(q22)[7] |
Negative for del(17p), del(11q), and del(13q) |
NA |
+i(5)(p10), del(7)(q31.32q36.1) |
Partial concordance |
|
26 |
PCM |
43,X,der(X)t(X;3)(p11.4;q13.1), del(1)(p21p31),del(2)(q24q32), del(4)(p15.1p15.3),psu dic(12)t(12;18)(p11.2;p11.2),-13,del(14)(q21q31),del(20)(p11.2),-22[5]/46,XX[15] |
Positive for del(17p) and t(4;14)
Negative for t(11;14) and t(14;16)
|
NA |
t(3;X)(q13.13;p11.4), dup(3)(q13.13q29), del(X)(p11.4p22.33), del(1)(p21.1p31.1), del(2)(q24.1q32.1), del(4)(p15.1p15.33), del(12)(p12.1p13.33), del(18)(p11.22p11.32), -13, del(14)(p21.3p31.3), t(4;14)(q16.3;q32.33), del(20)(p11.2),-22, del(17)(p13.1) |
Partial concordance |
|
27 |
PCM |
45,X,t(X;6)(p11.2;p25), der(14)t(11;14)(q13;q32), der(18)t(18;21)(q22;q21),-21[cp7]/46,XX[33] |
Positive for t(11;14)
Negative for del(17p), t(4;14), and t(14;16)
|
NA |
t(18;21)(q22.3;q21.1), del(18)(q22.3q23), del(21)(q21.1q21.3),dup(11)(q13.4q23.1) |
Partial concordance |
In two of the complex cases with partial concordance (S15 and S26), OGM failed to correctly detect SVs involving centromeric regions. Specifically, in S15, OGM accurately identified all three aneuploidies but missed the t(1;10)(q21;q11.2) associated with the derivative chromosome 10. Similarly, in S26, OGM accurately detected six deletions, two aneuploidies, one translocation, and an IGH::FGFR3 rearrangement but missed the t(12;18)(p11.2;p11.2) associated with the pseudodicentric chromosome 12. In another partially concordant complex case, S20, OGM failed to correctly detect SVs involving the telomeric region, specifically missing a microdeletion in the telomeric region of Xp22.33 and the P2RY8::CRLF2 fusion resulting from the microdeletion, identified only using the RNA fusion panel. This microdeletion was not captured during routine diagnostic testing using NGS CNV analysis.
In three other complex cases, S16, S25, and S27, OGM failed to correctly detect SVs in minor subclones. In S16, two deletions were detected correctly, whereas t(2;14)(q35;q13) and 14q13q32 gain associated with derivative chromosome 2, loss of chromosome 14, and del(13)(q13q22) were missed by OGM. These SVs were observed in only 16% (3/19 cells) of cases in CBA. In S25, one deletion and a gain of isochromosome, found in 18/25 cells in CBA, were accurately identified by OGM. However, OGM failed to detect one deletion that was identified in the minor subclone comprising 28% (7/25 cells) of cases in CBA. In S27, OGM correctly detected one unbalanced translocation and one aneuploidy, but missed two translocations, t(X;6)(p11.2;p25) and t(11;14)(q13;q32), and the IGH::CCND1 rearrangement. These SVs were part of the composite karyotype, which constituted 17.5% (7/40 cells) of cases in CBA. Finally, in one complex case (S24), only one balanced translocation was correctly detected by OGM, whereas loss of chromosomes 17 and X, gain of der(6;9)(p10;p10), and a marker chromosome were all undetected by both OGM and NGS CNV analysis, leading to a notable discrepancy as these SVs were observed in a substantial proportion of cells (i.e., 61% [11/18 cells]) in CBA.
DISCUSSION
We analyzed SVs in various hematologic malignancies using OGM and compared the results with those of routine diagnostic tests (CBA, FISH, RT-PCR, and an RNA fusion panel) to investigate their concordance. Overall, OGM showed concordance in 63% (17/27) of cases and partial concordance in 37% (10/27), with no cases of non-concordance. OGM correctly identified 76% (52/68) of total SVs, with specific concordance rates for each type of aberration as follows: aneuploidies (83% [15/18]), balanced translocations (80% [12/15]), unbalanced translocations (54% [7/13]), deletions (81% [13/16]), duplications (100% [2/2]), inversion (100% [1/1]), insertion (100% [1/1]), marker chromosome (0% [0/1]), and isochromosome (100% [1/1]). Compared with previously published results [
30], we observed a relatively low concordance rate, especially in complex cases. OGM showed limitations by detecting only part of the SVs or entirely missing some SVs. We analyzed the possible reasons for all discordant results and found they could be summarized as follows: i) SVs involving centromeric and/or telomeric regions, ii) SVs in minor subclones with low frequency likely below the detection sensitivity, and iii) SVs in samples with low mapping rate and coverage (
Table 3).
Table 3
Structural variations missed by OGM and probable cause of detection failure
|
Sample ID |
Diagnosis |
Structural variations missed by OGM |
Possible explanation for discordant results |
Comment |
|
12 |
CML-CP |
t(3;11)(p21;q13) |
Low map rate and coverage(map rate 24.5%, coverage 99.11×) |
|
|
13 |
CML-CP |
t(7;9)(q22;q34) |
Involvement of telomeric region |
|
|
14 |
PMF |
t(1;6)(q21;p21), gain of 1q21q44 |
Detection sensitivity |
Detected in 15% (3/20) of cells in CBA |
|
15 |
MDS-EB-2 |
t(1;10)(q21;q11.2) |
Involvement of centromeric region |
|
|
16 |
MDS-SLD |
t(2;14)(q35;q13), gain of 14q13q32, monosomy 14, del(13)(q13q22) |
Detection sensitivity orlow map rate and coverage(map rate 28.4%, coverage 87.75 X) |
Detected in 16% (3/19) of cells in CBA |
|
20 |
B-ALL |
Microdeletion in Xp22.33 |
Involvement of telomeric region |
|
|
24 |
MZBCL |
Monosomy 17, monosomy X, +der(6;9)(p10;p10), marker chromosome |
Low map rate and coverage(map rate 38.9%, coverage 98.85 X) |
Not detected by NGS CNV |
|
25 |
LPL |
del(16)(q22) |
Detection sensitivity |
Detected in 28% (7/25) of cells in CBA |
|
26 |
PCM |
t(12;18)(p11.2;p11.2) |
Involvement of centromeric region |
|
|
27 |
PCM |
t(X;6)(p11.2;p25), t(11;14)(q13;q32) |
Detection sensitivity |
Detected in 17.5% (7/40) of cells in CBA |
Highly repetitive regions, such as centromeric regions, short arms of acrocentric chromosomes, pseudo-autosomal regions (PARs), and telomeric regions, are often poorly covered by OGM because of missing labels and unreliable reference map data. Consequently, OGM may fail to detect SVs involving these regions, a well-known limitation [
14-
17]. However, as these regions may contain clinically relevant SVs, efforts should be undertaken to identify recurrent SVs in these areas. For instance, a microdeletion in the PAR of Xp22.33 and the
P2RY8::CRLF2 fusion caused by the microdeletion were missed by OGM in S20. A similar detection failure has been reported previously [
18]. Studies have reported
P2RY8::CRLF2 as a recurrent rearrangement in B-ALL that is associated with distinctive features and poor prognosis and may influence treatment choice [
31-
33]. The detection failure of recurrent SVs that have clinical significance by OGM is a critical problem that must be resolved.
For undetected SVs present in minor subclones in four cases (S14, S16, S25, and S27), we attribute the discrepancy to the detection sensitivity of OGM. These SVs were detected in a median of 16.75% of cells by CBA (range: 15%–28%). While OGM missed these subclonal SVs, it detected other SVs present in the same subclone. Additionally, some SVs were identified in only 15%–20% of cells using CBA among the concordant cases, indicating inconsistent detection sensitivity of OGM with respect to CBA results. Similar results have been reported previously [
16], and Rack,
et al. [
14] additionally performed interphase FISH and established that OGM detected SVs present in at least 15% of cells. However, we could not perform additional interphase FISH on these cases owing to a lack of samples.
For samples with discordant results attributed to a low mapping rate and coverage, the mapping rate was below 60%, and coverage was below 100×. Many other samples had a mapping rate below 60%, but the distinguishing factor was coverage. Other samples with a low mapping rate had a coverage above 100× (median 174.16×, range: 102.65–277.26×), whereas the discordant samples had a coverage below 100× (median 98.85×, range: 87.75–99.11×). This aligns with the Bionano Molecule Quality Report Guidelines that state that when the obtained mapping rate is significantly lower than the minimum desired mapping rate (i.e., <60%), extra depth can compensate for the low mapping rate. However, there was one exceptional case (S5) where OGM correctly identified all SVs despite a low mapping rate and low coverage.
Overall, the largest number of missed SVs per sample was four for samples S16 and S24. For S16, there are two possible explanations for the discordant OGM results: detection sensitivity or low mapping quality. Although S24 can be classified as a discordant case attributable to a low mapping rate and coverage, the SVs detected using CBA may represent a culture selection bias because, similar to the OGM results, NGS CNV results revealed no SVs.
Despite the aforementioned limitations, OGM demonstrated many advantages in detecting SVs. First, OGM was able to detect different types of SVs simultaneously in a single test in many cases. Second, OGM clarified the boundaries/breakpoints of SVs observed in CBA owing to its higher resolution. Third, OGM identified
IGH rearrangements even when they did not result in chimeric transcripts. We included two PCM cases, S26 and S27, to assess the capability of OGM to identify these rearrangements, which are challenging to detect using RNA sequencing. OGM successfully detected
IGH::FGFR3 in S26 but failed to detect
IGH::CCND1 in S27. The detection failure in S27 was probably attributed to detection sensitivity, as mentioned earlier. For a reliable and accurate conclusion, more cases must be examined. Fourth, OGM found additional SVs, including submicroscopic SVs and novel gene fusions with potential clinical significance. In AML case S4, OGM revealed that the inserted segment in chromosome 9 was a
MYC amplification, which implies a poor prognosis in AML [
34]. In B-ALL case S18, OGM newly detected del(12)(p13.31p12.1), del(17)(p11.2p13.3), dup(17)(p11.2q25.3), and
EP300::ZNF384 fusion. In a previous study,
EP300::ZNF384 was detected only by OGM in a B-ALL case [
14], and this aberration may influence the clinical outcome of B-ALL [
35]. In B-ALL cases S17 and S21,
SETD2,
CDKN2A/B, and
IKZF1 deletions were discovered by OGM, and all of these submicroscopic deletions have the potential to influence risk stratification, prognosis prediction, and patient management.
SETD2 deletions have been identified in various leukemias, and its loss has been associated with chemotherapy resistance [
36]. Similarly,
IKZF1 deletions are emerging as an important prognostic biomarker and are associated with resistance to tyrosine kinase inhibitors in B-ALL. Co-occurring
CDKN2A/B and
IKZF1 deletions are associated with worse outcomes [
37].
Collectively, our findings indicate that OGM can play a role in revealing many clinically significant SVs. Furthermore, in CML-CP case S11, OGM helped discover a novel gene fusion, ARL2-SNX15::CABIN1, which is a key strength of this technique. Identifying such fusions using OGM could pave the way for further investigations that may lead to clinically significant discoveries in the pathogenesis, diagnosis, prognosis, and treatment of hematological malignancies.
In addition to the intrinsic limitations of the OGM technology, this study had additional limitations. Its retrospective design is a primary limitation, and future studies with a prospective design are essential to validate the findings and establish the clinical utility of OGM. Moreover, the study assessed only a limited number of samples and variants, warranting future research involving larger cohorts with sufficient sample size and the analysis of a broader range of SVs for each specific hematologic malignancy diagnosis. Third, not all conventional methods (karyotyping, FISH, RT-PCR, and RNA fusion panel analysis) were consistently performed in all cases during routine diagnostic work-up, limiting the accuracy of the evaluation. A more comprehensive assessment could be achieved by consistently applying all conventional methods to every sample included in the study. Finally, as this study primarily focused on evaluating concordance, further studies are necessary to evaluate the performance characteristics of OGM, including sensitivity, specificity, and predictive values.
In conclusion, we assessed the concordance between OGM and traditional diagnostic methods for detecting SVs in hematologic malignancies, revealing a certain level of concordance. However, limitations were observed in detecting SVs involving centromeric and/or telomeric regions, minor subclones with low frequency, and samples with low mapping rates and coverage. Despite these limitations, OGM holds substantial value in identifying variants undetectable by conventional methods and discovering novel variants, suggesting its potential utility in comprehensively profiling SVs in routine diagnostics of hematologic malignancies. The identification of novel variants through OGM may yield clinically significant insights into the pathogenesis, diagnosis, prognosis, and treatment of hematological malignancies. However, for the implementation of OGM in clinical practice, further validation through prospective studies with larger cohorts and ongoing technical and analytical enhancements to address current limitations is imperative.


 PDF
PDF Citation
Citation Print
Print



 XML Download
XML Download