

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Intrahepatic cholangiocarcinoma (iCCA) is the second most common primary liver carcinoma (PLC), following hepatocellular carcinoma (HCC).1 PLC demonstrates a histological spectrum and continuity relative to its cell of origin.2 The human liver has three epithelial cell types that can give rise to cancer, namely, hepatocytes, cholangiocytes, and hepatic progenitor cells (HPCs). Heterogeneity is a distinguishing feature of iCCA and combined HCC-cholangiocarcinoma (CCA) as HPCs are the origin of their carcinogenesis.3 This makes them difficult to distinguish, by both imaging and pathological interpretation. An accurate diagnosis is, however, essential since completely different treatments are applied depending on the PLC subtype. In particular, iCCA harbors actionable mutations in more than 50% of cases.4,5
With all of this in mind, this review focuses on iCCA as it faces a turning point in treatment strategy, and raises the importance of diagnostic accuracy and the essential role of the pathologist.
Go to : 
THE BILIARY TREE
CCA arises from the biliary tree, which itself originates from the canal of Hering, and terminates at the choledochus. The biliary tree gathers bile produced by hepatocytes and drains the bile to the duodenum via the Vater papilla. Its structure, therefore, demonstrates that of a tree, and the liver contains its branches and leaves. Cholangiocytes are the lining epithelia of the biliary tree, and CCA originates from the cholangiocytes. Importantly, cholangiocytes demonstrate different phenotypes depending on their location (Fig. 1). The most distinct difference is seen in the smallest bile duct, named the ductule, and the large bile duct (i.e., hepatic ducts, and the extrahepatic bile duct). The cholangiocytes located in the large bile duct show similar features to the epithelia of the pancreatic duct, such as mucin-containing cylindrical cells. In contrast, the cholangiocytes of the ductule are small, cuboidal shaped cells with no mucin-production.
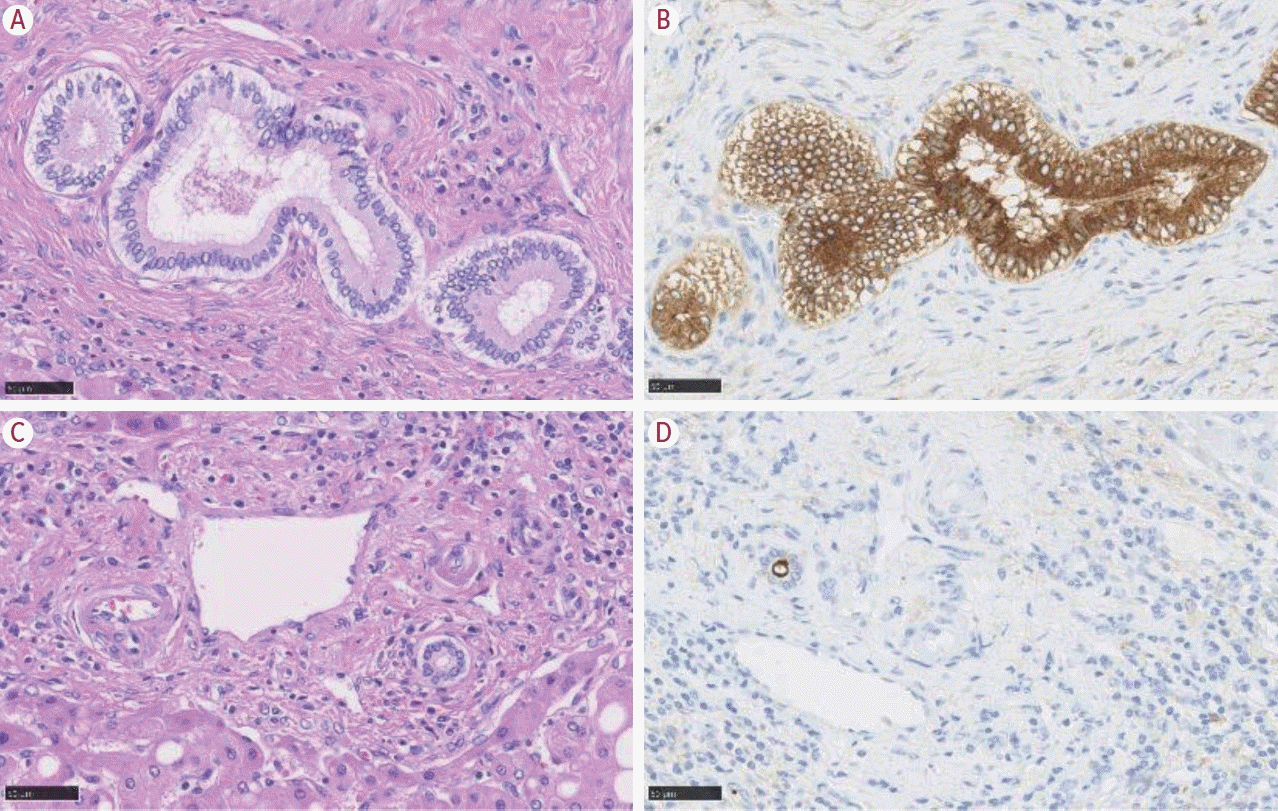 | Figure 1.Heterogeneity of cholangiocytes. The cholangiocytes are mucin-producing, cylindrically shaped cells in the large bile duct (A). In contrast, the small duct contains mucin-negative, cuboidal cholangiocytes (C). The different phenotype is clearly visible with epithelial membrane antigen (EMA) staining. Apical staining in the small duct (D), and cytoplasmic expression in the large bile duct (B) (A, B: hematoxylin- eosin; C, D: EMA immunohistochemistry; A-D: 200×; modified from Komuta12 with permission).
|
As cancer is known to preserve the features of its cell-of-origin, it is quite understandable that CCA can present with a different phenotype based on its location. This can influence, not only the pathological and morphological aspects of the tumor, but also clinical and genetic alterations. This is clearly illustrated by the 5th World Health Organization (WHO) classification, which introduces new subtypes, namely the large and the small duct type.6 This will be discussed later.
Go to :
CCA
Overview and limitations of the anatomy-based classification
CCA is classified as iCCA, perihilar, and distal (dCCA), based on location. In brief, iCCA occurs distal from the second-order branches of the biliary tree, while perihilar CCA is from the right and/or left hepatic duct and/or at their junction, and dCCA arises from common bile duct, especially below the insertion of the cystic duct. The distance between the secondary branches of the biliary trees and cystic duct is around 2-3 cm, thus it is not practical or precise to categorize CCA based on location as iCCA is often diagnosed at an advanced stage. In addition, the predominant location of the tumor is not always at the site of origin, and small duct-derived iCCA can occur in a perihilar location due to the three-dimensional biliary structure. This is one of the possible reasons that CCA is described as a heterogenous tumor, as perihilar CCA may contain iCCA, and vice versa. To overcome this dilemma, we previously categorized CCA based on its histological features, and were able to characterize CCA into two clear groups.7 In brief, we categorized CCA based on the presence of histological diversity; CCA with histological diversity was defined as mixed-iCCA, while iCCA without heterogeneity was defined as muc-iCCA. Muc-iCCA presented with a similar profile to hilar CCA (Klatskin tumor), while mixed-iCCA was more similar to cholangiolocellular carcinoma (CLC), which is considered to be of HPC origin (Fig. 2). In addition, this data illustrated that around 30% of CCAs are misclassified7 by anatomical-based classification. The anatomy-based CCA classification is, however, useful for assessing the operative approach when the tumor is resectable. This indicates that the classification method should be chosen based on the aim, as a single classification approach cannot cover every aspect.
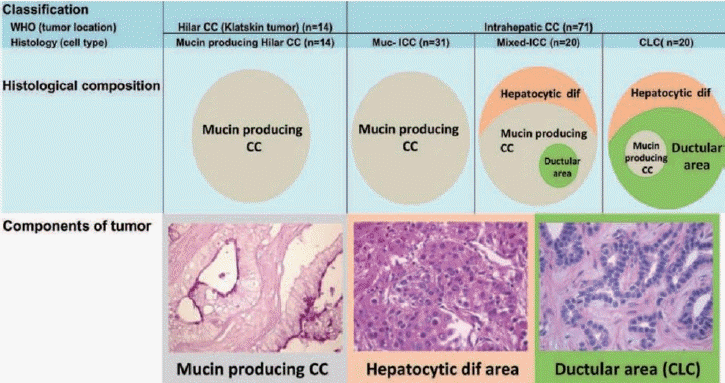 | Figure 2.Histological variation of cholangiocarcinoma (CCA). Hilar CC shows mucin producing CCA, similar to those from intrahepatic cholangiocarcinoma (iCCA) with mucin producing features. In contrast, tumor heterogeneity is a feature of cholangiolocellular carcinoma (CLC) and mixed-iCCA. Reused from Komuta et al.7 with permission. WHO, World Health Organization; CC, cholangiocarcinoma; Muc-iCCA, mucin-production intrahepatic cholangiocarcinoma.
|
Go to :
iCCA
Etiology-based classification
iCCA is often associated with biliary diseases, such as primary sclerosing cholangitis (PSC), hepatolithiasis, and parasites (liver fluke).1 Recently, however, chronic hepatitis due to viral hepatitis, and metabolic syndrome, have also been found to be risk factors for iCCA.1 Interestingly, the etiology has a close relationship with the subtype of iCCA: the large duct type often arises in PSC, hepatolithiasis, and parasitic infection, which all cause chronic inflammation. In contrast, viral hepatitis and metabolic syndrome are more common in the small duct type.3
Macroscopic classification
Macroscopically, iCCA is categorized into mass-forming (MF), periductal infiltration (PI), and intraductal-growing (IG) types.1 PI + MF is a tumor that initially presents with a PI pattern that subsequently invades into neighboring liver parenchyma and forms a mass. Importantly, the PI + MF type is often predominantly MF and the PI area is not always visible. Therefore, it is important to know that MF may comprise two types, which are pure MF and pseudo MF, with the PI component not visible. The IG type is less frequent compared with the other subtypes, and presents as a papillary and/or polypoid structure growing into the lumen. This type could also present as a mixed form, IG + MF, that is a MF type iCCA associating with intraductal extension (IG) pattern.8
Histological classification
iCCA has been treated as a single tumor entity for a long time, however, as of the 5th WHO classification, iCCA has been divided into two subcategories: the large and the small duct type (Table 1). This classification is based on the heterogeneity of cholangiocytes, as described above. Even though this classification depends solely on the tumor morphology, it correlates clearly with the clinical and molecular features. From the pathologist’s perspective this is important as a histological subclassification can predict genetic alterations prior to a genetic test, or predict prognosis.
Table 1.
Clinicopathological comparison between small and large duct type iCCA
Reused from komuta [12] with permission.
iCCA, intrahepatic cholangiocarcinoma; PSC, primary sclerosing cholangitis; PI, periductal infiltrating; MF, mass-forming; IDH, isocitrate dehydrogenase; FGFR, fibroblast growth factor.
![]()
Large duct type
The large duct type is characterized by mucin-producing glandular features, which mimic the mucin-producing, cylindrical-shaped cholangiocytes of the large bile duct (Fig. 3). This subtype is thought to originate from the large bile duct, with the tumor location mainly in the perihilar area. In addition, as it arises from the large bile duct, the tumor exhibits a macroscopic PI pattern. The large portal tract comprises neural tissue and lymphatic vessels, and the tumor thus often demonstrates perineural and/or lymphatic invasion. As such, this type has a worse prognosis compared with the small duct type, which is understandable when considering the tumor’s aggressive pathology.
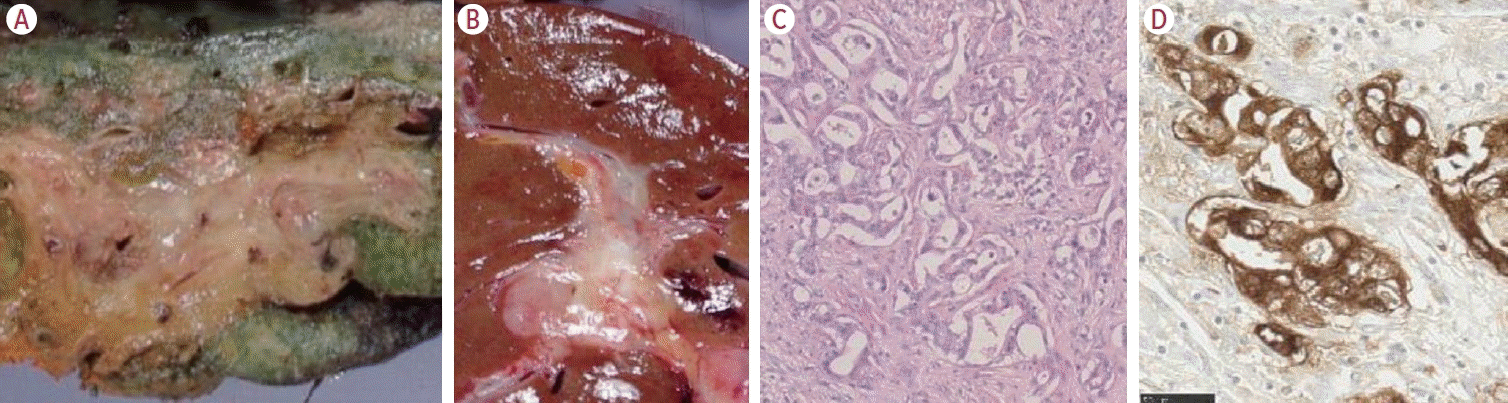 | Figure 3.The large duct type. Macroscopically, the tumor demonstrates periductal infiltrating type (PI) (A), and PI and mass-forming type (B). The tumor exhibits a clear glandular structure with mucin production (C), and cytoplasmic expression with epithelial membrane antigen (EMA) staining (D) (C: hematoxylin-eosin; D: EMA immunohistochemistry; C: 40×; D: 100×; modified from Komuta12 with permission).
|
Small duct type
The small duct type exhibits contrasting features to those of the large duct type (Fig. 4). The tumor appears as a small uniform gland, or irregular glandular structure with a narrowing lumen. Cellular atypia is mild, and often described as ‘innocent-looking’. The tumor stroma can be hyalinized mature stroma, or edematous with marked inflammatory infiltrations. Perineural and lymphatic invasion are less compared with the large duct type, and postoperative prognosis is thus better, especially when the tumor size is less than 3 cm. Macroscopically, all small duct type tumors are mass-forming: one of the key aspects for clinical distinction of the large and the small duct type.
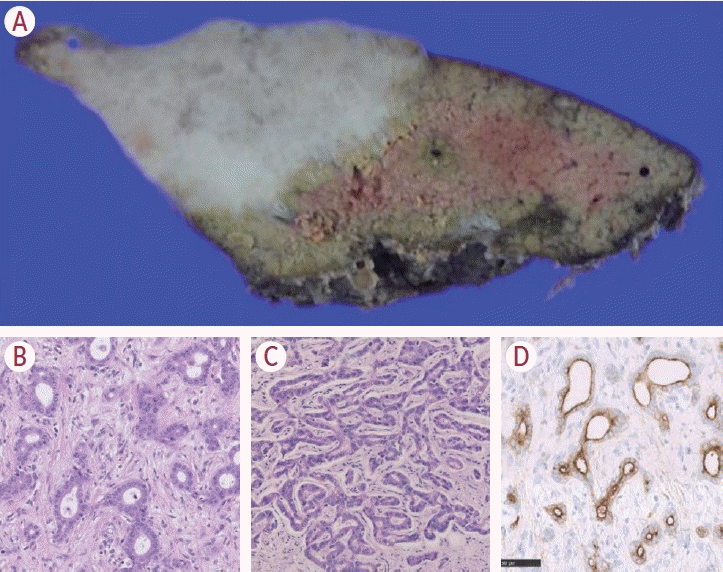 | Figure 4.The small duct type. The tumor shows mass-forming aspect (A), often arising from chronic liver diseases. The tumor is characterized by small uniform glandular structure (B) or ductular reaction-like structure (C) without mucin production. Epithelial membrane antigen (EMA) staining shows an apical expression pattern (D) (B, C: hematoxylin-eosin; D: EMA immunohistochemistry; B-D: 40×; modified from Komuta12 with permission).
|
Mixed type
A certain proportion of iCCA demonstrate mixed features comprising aspects of both the large and small duct type. Studies have shown that the mixed type is more closely aligned with the small duct type than the large duct type, highlighting the importance of correctly identifying the small duct type for iCCA subcategorization.
Genetic alteration-based classification
Approximately 50% of iCCAs have actionable mutations that can be treated with commercially available medications. The most frequently observed alterations are isocitrate dehydrogenase (IDH)-1 and IDH-2 mutations (18.3%) and fibroblast growth factor receptor 2 (FGFR2) fusions (11.6%), followed by ERBB2 (5.1%), and BRAF (5.0%).9 The Food and Drug Administration (FDA) and European Medicines Agency (EMA) have approved IDH and FGFR2 inhibitors, whereas Japan has only approved the FGFR2 inhibitor at this time. Clinical trial data indicate a favorable prognosis in iCCA patients treated with these inhibitors.10,11 Again, this highlights the importance of correctly diagnosing iCCA, including the correct subtype, as iCCA with the MF type and the small duct type are strongly correlated with these genetic alterations.12
Immunotherapy is also available for iCCA patients, for example in combination with the first line chemotherapy (gemcitabine + cisplatin), or with the use of pembrolizumab in microsatellite instability (MSI)-high patients. Unfortunately, the incidence of MSI-high iCCA is very low, and the relationship between iCCA subtypes and MSI status is not clear.
Prognosis
Following curative resection, the prognosis was better in the small duct type compared to the large duct type.13 In addition, FGFR fusion appeared to correlate with a better prognosis, most likely due to the fact that FGFR fusion is seen in the small duct type. Likewise, KRAS/BRAF mutations correlated with a worse prognosis as these mutations are associated with the large duct type iCCA.14
Go to :
CLC
CLC is a unique tumor that is believed to originate from HPCs due to its histological diversity.15 According to the WHO classification, CLC is defined as a tumor comprising ‘typical’ small duct features, such as ductular reaction-like features, in more than 80% of the tumor.6 In other words, CLC is considered as a distinct phenotype of small duct type iCCA. CLC often presents with hepatocytic differentiation, which is why the 5th WHO classification has recategorized CLC into combined HCC (cHCC)-CCA when hepatic differentiation is evident. CLC without hepatocytic features is still classified as a biliary tumor iCCA subtype. There is thus some confusion regarding tumor categorization since there is no clear definition of ‘hepatocytic features’, including by immunohistochemistry. There are several immunohistochemical markers available for hepatocytic differentiation. The most commonly used immunohistochemical markers are HepPar-1 and Arginase-1, but their sensitivities differ.16 Unfortunately, there are no guidelines on which, and how many, markers must be used to determine hepatocytic differentiation. Therefore, further investigation in the categorization of this tumor is required to standardize these markers for this purpose.
Go to :
DIAGNOSIS
There is no doubt that a precise diagnosis is crucial in iCCA in terms of treatment choice. To improve diagnostic accuracy, there are two issues that we have to keep in mind: the exclusion of metastatic tumors, and the PLC subtypes.
Differential diagnosis between iCCA and metastatic adenocarcinoma to the liver
iCCA is an adenocarcinoma that is the most frequent histological subtype of metastatic liver tumors. In addition, metastatic adenocarcinoma is more frequent compared to the primary adenocarcinoma, iCCA, and distinguishing them is crucial as their treatment and prognosis are different. Tumor number is not always helpful in identifying them. Therefore, a tumor biopsy is an important factor in the diagnosis. The diagnosis is usually made using appropriate immunohistochemical markers selected following observation of the morphological features of the tumor. For example, CDX2 is used to differentiate colorectal cancer (Fig. 5), GATA3 is used to differentiate breast cancer, and TTF-1 is used to differentiate lung cancer. However, a comprehensive diagnosis, including clinical and imaging information, is essential to distinguish a primary tumor from a metastasis.
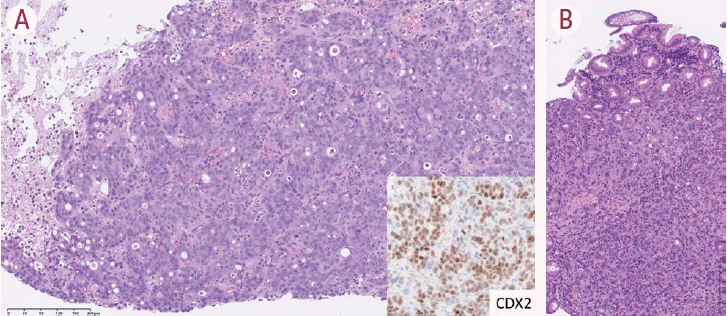 | Figure 5.A case of metastatic colon cancer of the liver. A liver tumor biopsy from a patient with a liver tumor and a clinical diagnosis of intrahepatic cholangiocarcinoma (iCCA). The tumor biopsy shows trabecular/glandular structures composed of tumor cells with enlarged nuclei. Necrosis is also seen (A). Immunohistochemically, the tumor cells are positive for CDX2, indicating its colorectal origin (insert). A colonoscopy revealed a tumor whose morphology mimicked that of the liver tumor, confirming metastatic liver cancer from the colon (B) (A, B: hematoxylin-eosin stain, 40×; inset: CDX2, 100×).
|
Differential diagnosis between iCCA and the other tumor subtypes
As previously mentioned, PLCs demonstrate a histological spectrum and continuity relative to their cell-of-origin.2 Among the PLCs, cHCC-CCA is the most heterogeneous as it comprises both hepatocytic and cholangiocytic differentiation within the same tumor.17 These different histological features show transitional aspects, which is important when distinguishing a collision tumor from cHCC-CCA. cHCC-CCA may demonstrate cholangiocytic features that could be small duct type iCCA, or even the large duct type iCCA. Therefore, the distinction between them could be challenging in a biopsy specimen that only represents a small part of tumor. However, when combined with radiological imaging, a biopsy is still useful to contribute to the overall picture that may lead to an accurate diagnosis.18
Go to :
CONCLUSIONS AND FUTURE PERSPECTIVES
iCCA is a heterogeneous tumor that can be categorized macroscopically into an MF, PI, and IG pattern, and histologically subclassified into two types, the large and small duct type. It is important to recognize that the MF iCCA and small duct type iCCA subtypes frequently harbor actionable mutations, and this information can be very useful in terms of treatment decision and predicting the prognosis of iCCA patients. However, we have to keep in mind that diagnosing iCCA has several pitfalls, namely distinguishing iCCA from metastatic liver tumors and from other PLC subtypes. Pathologists have an important role to play in this regard, as they can differentiate between primary and metastatic tumors, and determine iCCA subtype.
iCCA occurs less frequently compared to HCC, and it is important that, via multicenter and international collaboration, we accumulate iCCA cases in order to examine the nature of iCCA.
Go to :
 XML Download
XML Download