

 PDF
PDF Citation
Citation Print
Print



Introduction
Materials and Methods
1. Study design and patients
Fig. 1.
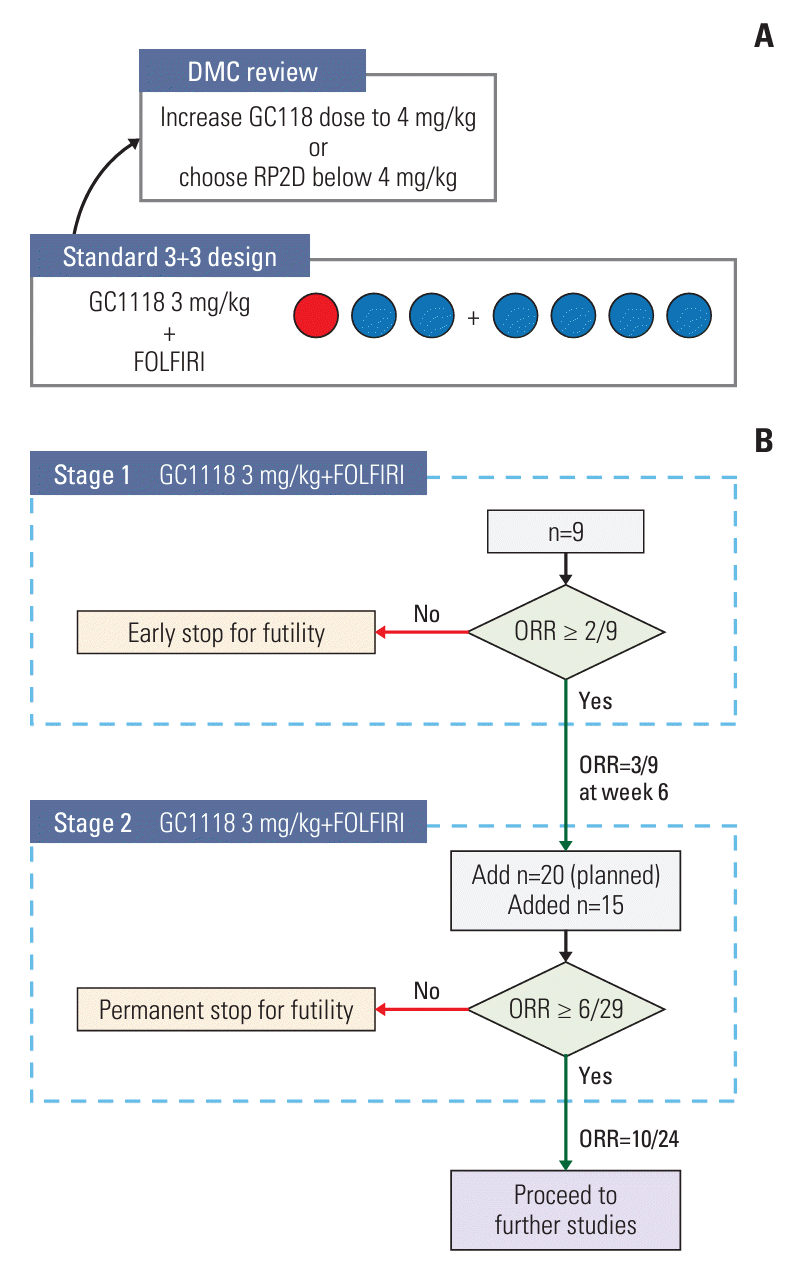
![]()
2. Treatment
3. Study endpoints and procedures
4. Statistical analysis
Results
1. Patients
Table 1.
| Phase 1b (n=7) | Phase 2a (n=24) | |
|---|---|---|
| Age (yr), median (min-max) | 58 (43-76) | 59 (44-72) |
| < 60 | 4 (57.1) | 13 (54.2) |
| ≥ 60 | 3 (42.9) | 11 (45.8) |
| Male sex | 3 (42.9) | 15 (62.5) |
| ECOG PS | ||
| 0 | 1 (14.3) | 13 (54.2) |
| 1 | 6 (85.7) | 11 (45.8) |
| Primary tumor site | ||
| Left-sided CRCa) | 5 (71.4)b) | 16 (66.7)c) |
| Right-sided CRCd) | - | 8 (33.3) |
| Biliary tract | 1 (14.3)e) | - |
| Breast | 1 (14.3) | - |
| Duration of disease (mo), median (range) | 70.3 (18.8-108.4) | 14.4 (2.6-42.2) |
| EGFR positivity in tumor tissue | 5 (83.3)f) | 24 (100) |
| Any KRAS, NRAS, or BRAF mutations in tumor tissue | 0g) | 0 |
| Any mutations detected in plasma ctDNA | 3 (42.9) | 4 (16.7) |
| EGFR mutation | 0 | 0 |
| KRAS mutation | 1 (14.3) | 0 |
| NRAS mutation | 0 | 2 (8.3) |
| BRAF mutation | 0 | 0 |
| PIK3CA mutation | 0 | 2 (8.3) |
| PTEN mutation | 2 (28.6) | 1 (4.2) |
Values are presented as number of patients (%) unless otherwise specified. CRC, colorectal cancer; ctDNA, circulating tumor DNA; ECOG PS, Eastern Cooperative Oncology Group performance status; EGFR, epidermal growth factor receptor.
b) Among five patients with left-sided CRC, one patient had rectal cancer and four had left-sided colon cancer,
c) Among 16 patients with left-sided CRC, 11 had rectal cancer and five had left-sided colon cancer,
d) Right-sided tumor was defined as a tumor originating from ascending colon to entire transverse colon,
![]()
2. Treatment exposure
Table 2.
![]()
Table 3.
![]()
3. Safety
Table 4.
![]()
Table 5.
![]()
4. Immunogenicity
5. Efficacy outcomes
Fig. 2.
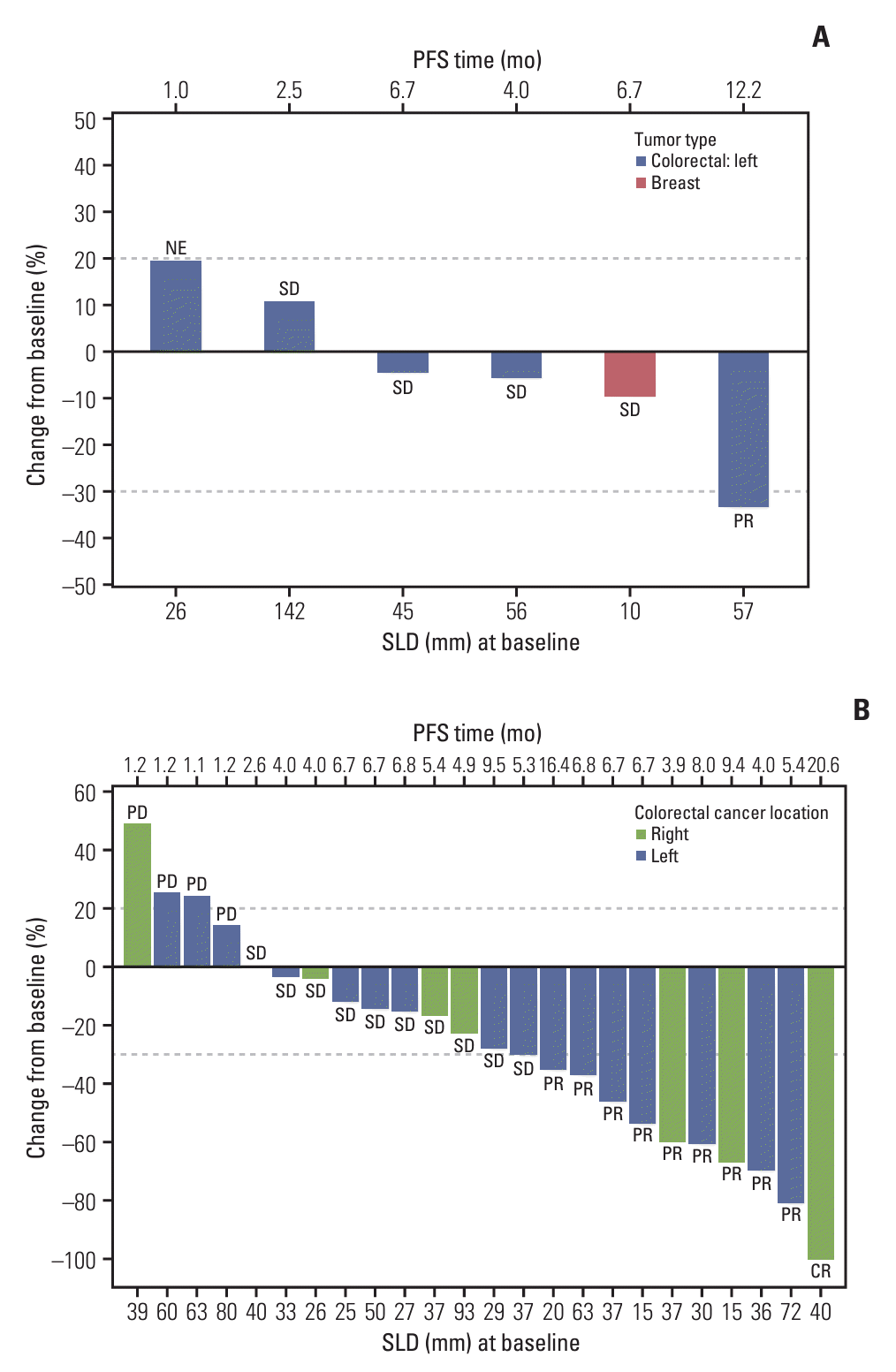
![]()
Table 6.
| Phase 1b (n=6) | Phase 2a (n=24) | |
|---|---|---|
| Complete response, n (%) | - | 1 (4.2)a) |
| Partial response, n (%) | 1 (16.7) | 9 (38.2)a) |
| Stable disease, n (%) | 4 (66.7) | 10 (41.1)a) |
| Progressive disease, n (%) | - | 4 (16.4)a) |
| Not evaluable, n (%) | 1 (16.7) | - |
| Objective response rate (%) (95% CI) | 16.7 (0.4-64.1) | 42.5 (23.5-62.0)a) |
| Disease control rate (%) (95% CI) | 83.3 (35.9-99.6) | 83.6 (51.8-97.2)a) |
| Progression-free survival (mo), median (95% CI) | 12.2 (2.5-NR) | 6.7 (4.0-8.0) |
| Progression-free survival (mo), RMST (95% CI) | 10.3 (6.9-13.6) | 7.1 (4.8-9.5) |
| Overall survival (mo), median (95% CI) | - | 25.4 (14.3-NR) |
| Time to respond (mo), median (95% CI) | -b) | 5.3 (1.3-NR) |
| Time to respond (mo), RMST (95% CI) | -b) | 5.8 (4.1-7.5) |
| Duration of response (mo), median (95% CI) | -b) | 6.8 (2.8-13.7) |
| Duration of response (mo), RMST (95% CI) | -b) | 7.9 (4.7-11.2) |
![]()
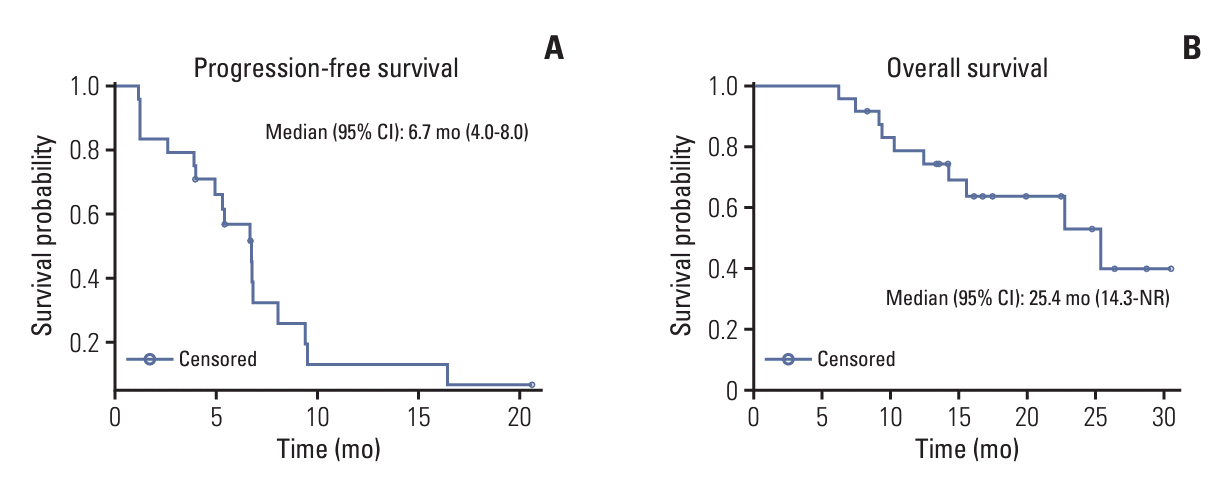
6. Exploratory outcomes
7. Pharmacokinetics
Table 7.
| Parameter | Single dosea) (n=7) | Parameter | Repeated doseb) (n=5) |
|---|---|---|---|
| Tmax (hr) | 1.5 (1.0-5.0) | Tmax,ss (hr) | 1.5 (1.0-5.0) |
| Cmax (μg/mL) | 46.5±10.6 (22.8) | Cmax,ss (μg/mL) | 69.7±13.3 (19.1) |
| Ctrough (μg/mL) | - | Ctrough (μg/mL) | 23.2±11.3 (48.7) |
| AUC0-168h (μg ‧ hr/mL) | 3,441.5±807.2 (23.5) | AUC0-168h,ss (μg ‧ hr/mL) | 6,704.7±2,128.1 (31.7) |
| AUCinf (μg ‧ hr/mL) | 4,603.8±1,772.0 (38.5) | AUCinf,ss (μg ‧ h/mL) | 10,574.5±2,562.0 (24.2) |
| MRTinf (hr) | 116.4±63.4 (54.5) | MRTinf,ss (hr) | 167.1±84.3 (50.4) |
| t1/2 (hr) | 81.8±46.7 (57.1) | t1/2 eff (hr) | 109.2±60.7 (55.6) |
| % AUCextrap | 20.7±15.7 (76.0) | % AUCextrap,ss | 35.3±17.8 (50.2) |
Values are summarized as arithmetic mean±standard deviation (coefficient of variation, %) except for Tmax, for which median (min-max) is presented. AUC0-168h, area under curve (AUC) from 0 to 168 hours post-dosing; AUCinf; AUC from time of dosing extrapolated to infinity; %AUCextrap; percentage of AUC obtained by extrapolation; Cmax, maximum serum concentration; Ctrough, serum concentration immediately prior to the next dose; MRTinf, mean residence time extrapolated to infinity; t1/2, elimination half-life; t1/2 eff, effective elimination half-life; Tmax, time to reach maximum serum concentration. Parameters obtained at steady state (ss) are denoted with ss.
a) Single-dose pharmacokinetic parameters were derived from the serum samples collected during the first cycle of the treatment (from seven hours before the first dosing to 120 hours after the dosing) and before the second dosing, which was administered on the eighth day of the first cycle,
b) For the repeated-dose pharmacokinetic analysis, blood samples were collected during the third cycle of the treatment (from 7 hours before the first dosing of the third cycle till 120 hours after the dosing) and before the second dosing, which was administered on the eighth day of the third cycle.
![]()
 XML Download
XML Download