

 PDF
PDF Citation
Citation Print
Print



Highlights
• MASH can lead to hepatic inflammation, fibrosis, cirrhosis and cancer.
• T2DM is a significant risk factor for fibrosis.
• Hepatocyte injury stimulates the activation of hepatic stellate cells into myofibroblasts.
• HCC in MASH can arise before cirrhosis has developed.
• Drugs to treat MASH are nearing reality and the first approval is likely soon.
INTRODUCTION
Obesity is linked to well recognized co-morbidities that comprise the metabolic syndrome, including type 2 diabetes mellitus (T2DM), hypertriglyceridemia, and hypertension [1]. However, the additional presence of steatotic liver, with or without inflammation and fibrosis, is often overlooked because these abnormalities typically do not have specific symptoms. This article reviews the fundamental mechanisms of fibrosis in steatotic (fatty) liver disease and their epidemiologic and mechanistic links to primary liver cancer, or hepatocellular carcinoma (HCC).
The terminology of steatotic liver disease has recently been standardized through an international effort [2]. Henceforth, the diseases formerly known as non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) are now termed metabolic dysfunction-associated steatotic liver disease (MASLD) and metabolic dysfunction-associated steatohepatitis (MASH), and these new terms are used in this review article. These new terms eliminate stigmatization by removing the word ‘fatty,’ by defining the disease by what is present instead of what is not present (e.g., non-alcoholic), and by accounting for the co-existence in some patients of liver disease-associated with both alcohol and metabolic dysfunction.
The escalating prevalence of obesity and T2DM has given rise to a staggering increase in MASLD and its more advanced form, MASH. A significant subset (approximately 20%) of individuals with MASLD progress to MASH, but the underlying factors that either promote or protect some individuals from progression of MASLD to either MASH, cirrhosis and HCC remain unclear. Overall, once cirrhosis is present there is an approximately 1.5% to 2% per year risk of developing incident HCC [3,4].
The distribution of MASH varies worldwide but is rising especially in Asia [5]. In Korea, the prevalence of MASLD is approximately 30% of the population [6,7], and is increasing in association with increasing T2DM [8]. MASH is estimated to occur in 10% of those with MASLD in Korea [9]. Worldwide, MASH in lean individuals (“lean MASH”) represents about 5% to 20% of the overall MASLD population [10,11]. Its prognosis is no better than in obese patients with MASH, and may be worse [12]. The mechanisms underlying lean MASH are not understood, but it is more prevalent among Asians, including Koreans [13].
Recent data indicates that the world is ill-prepared to handle the growing burden of MASLD, as few countries have implemented adequate public health measures to identify and manage the anticipated surge in patients while awaiting effective therapies [14,15] for a disease that significantly impairs health-related quality of life [16]. Thus, improving the awareness, diagnosis and treatment of MASLD and MASH are very high public health priorities.
METABOLIC DYSREGULATION IN MASH
MASH is associated with various pathways of metabolic dysregulation, both within the liver and systemically in extracellular tissues like adipose, muscle, and pancreas [17]. The liver, in particular, has altered signaling in pathways governing lipid homeostasis and carbohydrate metabolism. The result is an accumulation of fat from de novo lipogenesis, impaired lipolysis and hepatic export, as well as increased hepatic glucose uptake and impaired glucose utilization. These changes reflect an energy metabolism imbalance, with an excess of energy entering the liver compared to its capacity to oxidize or export these substrates. Multiple inputs from the microbiome, visceral adipose, muscle, the immune system, and the central nervous system influence these pathways.
Of all the co-morbidities associated with metabolic syndrome and steatotic liver disease, T2DM is single greatest risk factor for the development and progression of MASLD to MASH, and for accelerated progression of fibrosis in those with MASH [18,19]. Based on the strong association between T2DM and MASLD, the American Diabetes Association [20] and the American Association for the Study of Liver Diseases [21] have recommended screening all patients with T2DM for the presence of MASLD or MASH. Screening approaches vary based on the country and technologies available to clinicians, but typically include use of a non-invasive score (e.g., fibrosis-4 [FIB-4] or enhanced liver fibrosis [ELF]) combined with liver stiffness (Fibroscan, Echosens, Paris, France) [22,23]. Detection of MASLD or MASH among patients with T2DM is important in part because improved glycemic control may improve MASLD [24]. MASLD is also linked to increased adverse pregnancy outcomes in a Korean population [25].
UPSTREAM DRIVERS OF HEPATOCYTE INJURY IN MASH
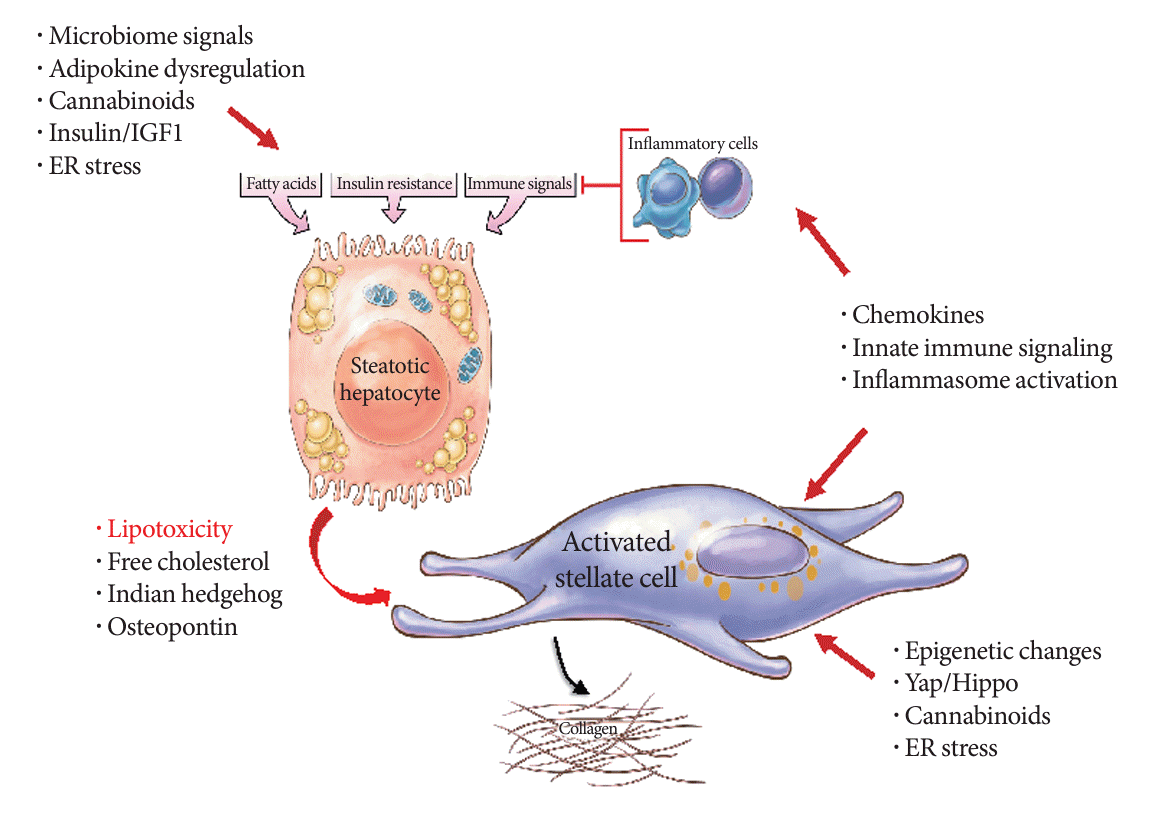
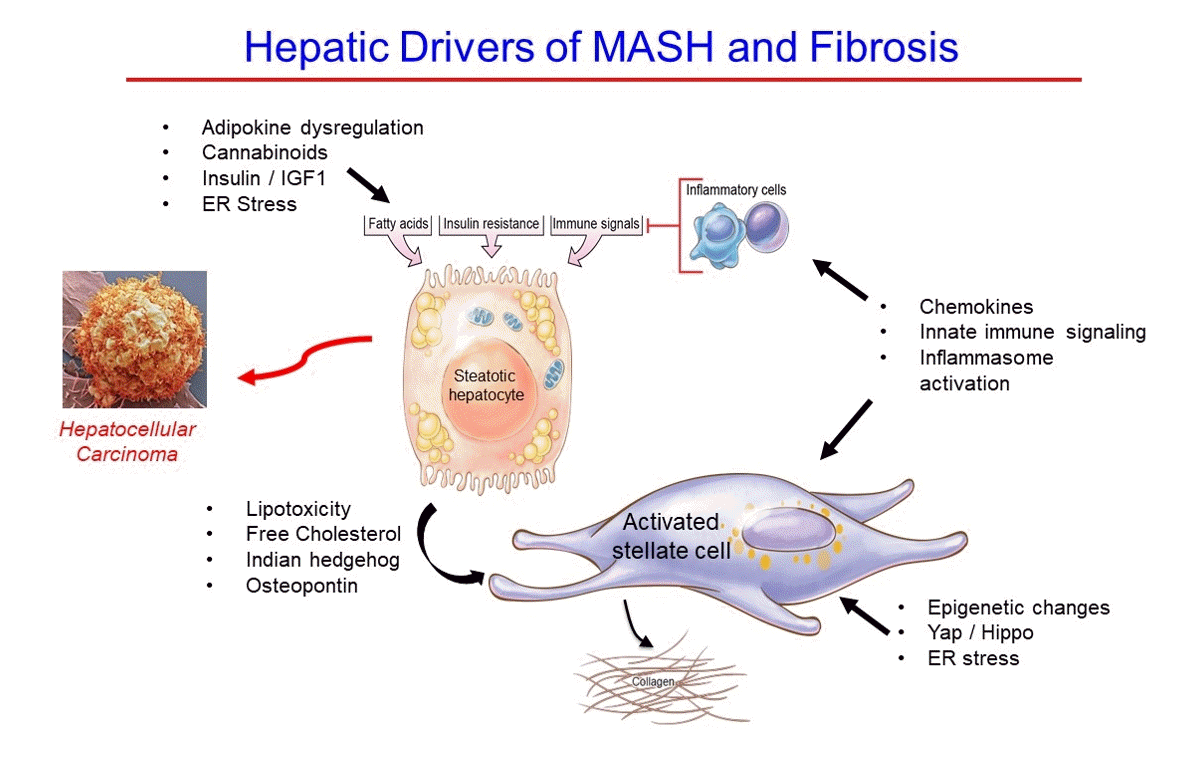
The factors initiating hepatocyte injury in MASH are not yet definitively identified, but several candidates have been proposed. These include dysregulated circulating adipokines and inflammatory molecules [26-29], elevated circulating insulin and insulin like growth factor (IGF), and signals from the gut microbiome, which exhibits abnormalities in MASH patients and animal models. With the advent of single cell RNA sequencing technologies, subsets of hepatocytes have been identified that may selectively promote the development of MASLD [30].
Gut dysbiosis, characterized by an unbalanced gut microbial community, is a compelling candidate for initiating and perpetuating MASLD, fibrosis, and HCC [31-33]. Dysbiosis can lead to increased gut permeability, enabling intestinal products to enter the portal vein, causing hepatocellular damage and sterile inflammation. Additionally, gut bacteria or their products may promote hepatocyte senescence and stimulate stellate cell activation. Intriguingly, certain gut bacteria in MASH have been found to generate ethanol, leading to liver injury, known as ‘autobrewery syndrome’ [34] although its overall contribution to MASH remains uncertain. Recent findings also suggest that the composition of the gut microbiome might influence the responsiveness of MASH and other disease treatments. Mouse models, although optimistic in predicting drug efficacy, have limitations due to genetic homogeneity and less complex microbiota compared to outbred mice, which better emulate human drug responses.
HEPATOCYTE DAMAGE AND FIBROSIS IN MASH
Upon hepatocyte injury, a cascade of signals involving intercellular crosstalk stimulates the activation or transdifferentiation of hepatic stellate cells into myofibroblasts, leading to increased fibrogenesis. These stellate cell-derived myofibroblasts play a central role in liver injury resulting from MASH and other causes of parenchymal cell damage [35,36]. They originate from the cardiac mesoderm/septum transversum during development and resemble pericytes in other tissues. These cells reside in the subendothelial space surrounding the sinusoid, where they can regulate sinusoidal blood flow [37]. With the advent of single cell RNA sequencing technologies, subsets of hepatocytes have been identified that may selectively promote the development of MASLD [30].
Genetic contributions to the pathogenesis of NAFLD (MASLD) are recognized [38-42]. A growing number of single nucleotide DNA polymorphisms, many of which are associated with lipid handling within liver cells (hepatocytes), have been identified as potential risk factors and therapeutic targets in NASH (MASH). The extent to which these genetic factors contribute to the onset and severity of MASH remains unclear, but they are likely among several multifactorial determinants of the disease. The importance of genetic contributions is further emphasized by the high frequency of MASH fibrosis among first-degree relatives [43,44], although it’s worth considering that familial clustering of the disease could also be influenced by a shared microbiome, particularly for those living in the same household. Although genetic variability is clearly a risk factor for the development of MASH and MASLD, genetics alone does not account for the increasing prevalence of the disease, and instead it is one of the factors that collectively enhance risk of disease or predispose patients to disease progression [45,46].
Therapeutic targets that have emerged are categorized based on their intervention points in the pathogenic sequence. Although not the primary focus of this review, general classes of therapeutics include those aiming to reduce fat accumulation in hepatocytes, improve insulin signaling and glucose homeostasis, counteract inflammation resulting from hepatocellular injury, decrease oxidative stress and restore metabolic and structural integrity of hepatocytes, as well as directly antagonizing fibrogenic signaling by activated stellate cells/myofibroblasts [47-49].
DIAGNOSIS OF MASLD AND FIBROSIS
The definitive diagnosis of MASLD and MASH still necessitates a liver biopsy of sufficient size and containing an adequate number of portal tracts to classify and stage the disease [50,51]. Initial studies primarily relied on the NASH Clinical Research Network (CRN), activity score, which comprises three main components: steatosis, lobular inflammation, and ballooning. Fibrosis is assessed separately using either a 0–4 (Brunt Kleiner score or SAF [steatosis, activity, and fibrosis] scores) or 0–6 scale (Ishak score). However, accumulating data have revealed limitations in the NASH CRN score, particularly due to significant sampling variability of its three features and challenges in defining and quantifying ballooning within a liver section. On the other hand, fibrosis has consistently emerged as the most crucial histologic feature predicting clinical events. This finding is supported by a significant longitudinal study from the NASH CRN, which followed almost 1,800 patients for 10 years and established the importance of fibrosis as a risk factor for death from any cause, hepatic decompensation, and HCC [52]. Consequently, both the disease’s pathologic scoring systems and therapeutic efforts are increasingly focused on accurately quantifying fibrosis and targeting pathways that enhance fibrogenesis or stimulate matrix degradation, directly or indirectly.
Recent data also support the superiority of digital pathologic methods in more accurately quantifying fibrosis content along a continuous scale [53-55]. As these methodologies are validated in longitudinal trials, they are likely to replace or complement conventional scoring systems. Furthermore, efforts are intensifying to replace liver biopsy altogether with non-invasive disease staging methods to broaden enrollment in clinical trials and improve patients’ eligibility for effective therapies once approved by regulatory authorities such as European Medicinces Agency or U.S. Food and Drug Adminstration.
FIBROSIS AND MASH
Fibrosis plays a pivotal role in determining clinical outcomes in MASH, making it crucial to delve into its pathogenesis in greater detail (Fig. 1). For decades, the activation of hepatic stellate cells has been recognized as a central event in fibrosis development [56,57]. Typically, these activated stellate cells express alpha smooth muscle actin and various cell surface and intracellular molecules that collectively promote their fibrogenic behavior. Recent advancements in single cell sequencing have shed light on stellate cell heterogeneity, revealing diverse phenotypes beyond the conventional ‘activated’ state [58]. This newfound understanding suggests the existence of stellate cell subtypes, including those previously activated but inactivated yet ‘primed’ to reactivate quickly after repeated liver injury [57,59]. Additionally, senescent hepatic stellate cells stimulated by microbiome signals adopt a pro-inflammatory senescenceassociated secretory phenotype that contributes to fibrosis progression. Targeting these senescent subpopulations through therapeutic approaches, such as CAR-T cells, has shown promise in improving fibrosis and injury in experimental MASH models in mice [60]. There remain other stellate cell subtypes whose functions are yet to be fully elucidated [58], but they likely contribute to the regional and individual differences observed in fibrosis extent and rate among MASH patients.
Stellate cell heterogeneity may also have significant implications for HCC pathogenesis [58,61]. Studies utilizing single cell sequencing have identified stellate cell subpopulations that either promote or inhibit HCC, depending on their secretory and genetic phenotypes [62]. These findings help resolve long-standing debates and contradictory observations regarding whether fibrogenic cells hinder tumor growth by encapsulating cancer cells, or promote tumor development through direct stimulation of cancer cell growth. The complexity of the tumor microenvironment is further highlighted by these discoveries, as it involves not only immune cells but also stromal cells, including activated stellate cells.
An important feature of advanced MASH fibrosis, not previously recognized, is the expansion and physical elongation of hepatic stellate cells as the disease progresses. These features help stellate cells develop a dense network of autocrine cell-cell interactions driven by a unique repertoire of ligand receptor combinations [63]. This insight suggests that as fibrosis advances, therapeutic targets evolve, potentially necessitating different drugs than those used when the disease primarily exhibits fat and inflammation with less fibrosis. This ‘cold’ fibrosis stage, characterized by autocrine interactions [64], likely explains why fibrosis continues to progress in advanced MASH patients, even after their livers have lost fat and other classic histologic features associated with MASH.
Understanding the intricate pathogenesis of fibrosis in MASH is of utmost importance, as it opens up new possibilities for targeted therapies that can effectively halt disease progression and improve patient outcomes. The emerging knowledge of stellate cell heterogeneity and its impact on HCC development offers promising avenues for future research and the development of innovative treatment approaches.
MASH-HCC: GENERAL FEATURES AND MASH-SPECIFIC DRIVERS
The incidence of MASH-HCC is on the rise globally, although its rates vary significantly across different regions [3,65]. Notably, MASH-related HCC has the fastest-growing rate among various chronic liver diseases and HCC etiologies. One distinctive clinical feature of MASH-HCC is its higher tendency to develop before cirrhosis is established. Roughly one-third of MASH-HCC cases arise in non-cirrhotic livers, whereas 95% of HCC cases associated with viral hepatitis occur in cirrhotic livers [3,66,67]. Nevertheless, cirrhosis remains the most significant risk factor for HCC in MASLD, emphasizing the importance of efforts to regress fibrosis in potentially protecting patients from liver cancer in the context of MASH.
Numerous features of MASH contribute to the unique clinical behavior of HCCs arising in this disease. Obesity, in particular, is a prominent risk factor for various cancers, including liver cancer. Obesity leads to chronic inflammation, heightened oxidant stress, DNA damage, and genomic mutations [26]. Additionally, it is associated with increased levels of mitogenic signals such as IGF and hepatocyte growth factor, as well as dysregulation of adipokines. The interplay between these factors, combined with potential genetic determinants specific to HCC risk and alterations in the gut microbiome, creates a dysregulated tumor microenvironment where HCC emergence may no longer depend solely on cirrhosis, although advanced fibrosis is still typically present. Obesity-induced systemic immune alterations, including heightened Th17-related inflammation, further impact the liver’s immune environment [26].
Recently, various molecular differences between MASH-HCC and non-MASH-HCC have been identified [65]. Of particular interest is the role of linoleic acid accumulation, leading to increased reactive oxygen species, cluster of differentiation 4 (CD4) T-cell depletion, auto-aggressive CXC motif chemokine receptor 6 (CXCR6) CD8 T-cells, metabolic reprogramming, and heightened DNA damage [27,68]. These changes are exacerbated by chronic dyslipidemia, endoplasmic reticulum stress, and other immune inflammatory changes associated with obesity.
One crucial consequence of the MASH-HCC-specific changes in hepatic immunity is the reduced efficacy of immunotherapy regimens that utilize checkpoint blockade [68]. Hepatic stellate cells may play a role in immunotolerance and resistance to checkpoint inhibition, thereby linking fibrogenic responses directly to a dysregulated immune microenvironment. While the success of some checkpoint inhibitors in treating primary HCC and reducing recurrence in MASH is promising, this area is rapidly evolving, and many questions remain unanswered.
FIBROSIS REGRESSION IN MASLD: CLINICAL FINDINGS AND UNDERLYING MECHANISMS
The development of highly effective therapies to either suppress hepatitis B, or cure hepatitis C, have created opportunities to unearth delivers and the alternate capacity to regress scar. In particular, long term suppression of hepatitis B virus (HBV) leads to marked diminution in advanced cirrhosis [69]. Similarly, curative therapies for hepatitis C virus (HCV) have again revealed the capacity of the healing liver to resorb the scar [70].
There are now also data suggesting that fibrosis is reversible in patients with MASH, primarily those who have undergone successful bariatric surgery associated with significant weight loss [71]. These patients have reduced fibrosis stage and improved overall outcomes for years after bariatric surgery [72]. Of note, however, cirrhosis associated with MASH may not be as reversible as in HBV and HCV, since a recent study indicates that cirrhosis persists if bariatric surgery is conducted after the disease has reached this advanced stage [73]. Bariatric surgery in MASLD has also been linked to reduced risk of HCC [74], which in part may be determined by underlying gene signatures of the disease that predict risk of cancer [75].
The mechanisms underlying fibrosis regression in any human liver disease remain understudied and unclear. Many years ago, sources of proteolytic enzymes or defined based on cellular and molecular methods, and focused on hepatic macrophages as a primary source of collagenases and enzymes that degrade other components of the extracellullar matrix [76]. More recently, subsets of macrophages, especially in Ly6Clo macrophages in mouse, have been implicated [77]. However, the overall activity of proteolysis is regulated not only by the amount of proteases, but also by the associated activity of metalloproteinase inhibitors or tissue inhibitors of metalloproteinases, as well as the localization of these enzymes relative to accumulating scar. To date, we lack a cohesive understanding of the cellular sources and disease-associated proteases that degrade scar. It is anticipated that with the availability of single cell methodologies, the mechanisms underlying scar degradation will be clarified. This is important, because such information could provide critical clues to develop novel therapies selectively amplifying scar degradation.
In summary, understanding the pathogenic features of MASLD and MASH-HCC is a top priority in hepatology. Recent advances have been significant, but there are still numerous unanswered questions that future studies must address. Some of these include:
1. Identifying the specific signals that explain why only a fraction of MASLD patients to develop MASH. Are genetics, the microbiome, diabetes, lipotoxicity, immune dysregulation, or a combination of these factors involved?
2. Considering whether MASH is a single disease with different subgroups, each with predominant pathways or abnormalities like insulin resistance, lipotoxicity, microbiome, or immune system dysfunction.
3. Exploring whether metabolic, anti-inflammatory, or antifibrotic therapies alone will suffice or if combination therapies are necessary to improve outcomes and reduce HCC in the majority of patients. Currently, even successful therapies benefit only a minority of patients, underscoring the need for further research.
Investigating and understanding these aspects of MASH and MASH-HCC will pave the way for more effective treatments and management strategies in the future. In particular the impact of T2DM on MASLD progression, mechanisms of fibrosis, and risk of HCC is not fully understood. Greater integration of expertise from endocrinology and hepatology will accelerate progress and improve outcomes for patients with metabolic syndrome and steatotic liver disease.
 XML Download
XML Download