

 PDF
PDF Citation
Citation Print
Print



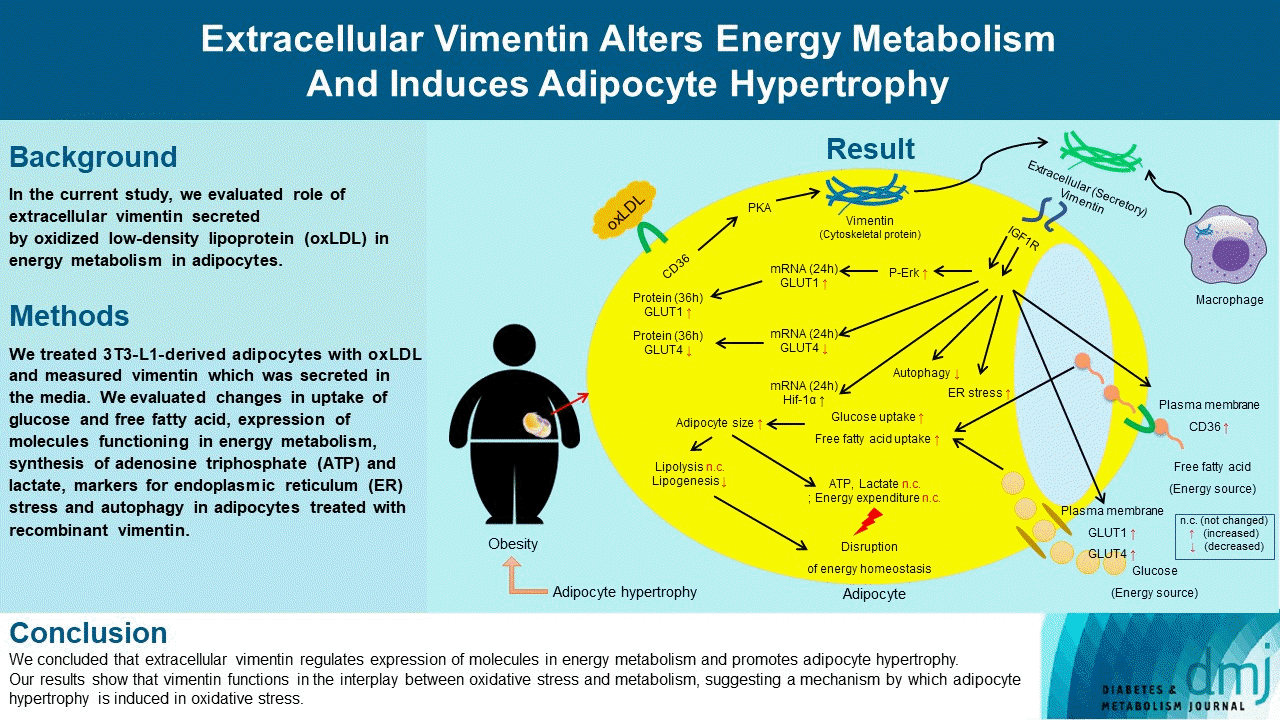
Highlights
• Extracellular vimentin is secreted by oxidized LDL in adipocytes.
• Extracellular vimentin increased ER stress and inhibited autophagy in adipocytes.
• It altered adipocyte energy metabolism, promoting adipocyte hypertrophy and obesity.
• Extracellular vimentin may mediate inflammation-obesity crosstalk.
INTRODUCTION
Obesity is a global health issue associated with a large number of disorders including diabetes and cardiovascular diseases [1]. Abdominal obesity is a significant risk factor for metabolic syndrome and visceral adiposity is coupled with oxidative stress [2].
Oxidative stress in obesity is driven by many factors. Intracellular glucose overload leads to excessive activation of glycolytic pathway and tricarboxylic acid (TCA) cycle, which contribute to production of reactive oxygen species (ROS). Increase in plasma free fatty acids (FFAs) also stimulates generation of ROS [3].
Inversely, oxidative stress is known to promote adiposity. Oxidative stress accelerates clonal expansion of preadipocytes [4]. However, effects of oxidative stress in adipocyte energy metabolism, a determinant of adiposity, has not been extensively studied. Therefore, there is a need for research that evaluates how oxidation products affect adipocyte function and energy metabolism to define pathogenesis of obesity.
Oxidized low-density lipoprotein (oxLDL), a marker of oxidative stress is positively associated with adiposity and insulin resistance [5]. However, there have not been many studies for metabolic changes of adipose tissue with high concentration of oxLDL. Adipose tissue serves as a major storage for excess energy and an endocrine organ synthesizing many compounds regulating metabolic homeostasis [6]. Previous study reported that oxLDL activates nuclear factor κB, inducing inflammation in adipose tissue. Adipose tissue inflammation is a crucial risk factor for obesity-associated insulin resistance and type 2 diabetes mellitus (T2DM) [7].
In inflammatory milieu as obesity, adipocytes become hypertrophic and limitation of oxygen diffusion induces adipose tissue hypoxia. Adipocyte hypertrophy increases cellular stress, decreases metabolic flexibility and induces diabetes. In addition, adipocyte hypoxia induces adipocyte death and inflammation [8]. However, mechanism of how adipocyte hypertrophy occurs in adipose tissue inflammation has not been clearly defined.
Vimentin is a type III intermediate filament protein and is mainly expressed in cells of mesenchymal origin including macrophages and adipocytes. Vimentin surrounds lipid droplets (LDs) in adipocytes [9]. Accordingly, vimentin deletion inhibits adiposity. Previously our group revealed that vimentin null mice fed a high fat diet showed less adiposity and improved glucose tolerance with higher levels of serum triglyceride (TG) and FFAs. We found that adipocytes from vimentin null mice showed less plasma membrane expression of CD36 and glucose transporter type 4 (GLUT4) compared to controls [10].
Vimentin is secreted extracellularly by pro-inflammatory cytokines in activated human macrophages [11]. Our group reported that oxLDL induces vimentin secretion and extracellar vimentin promotes inflammatory cytokine release in macrophages [12]. In addition, our group [12] and Gong et al. [13] found that serum levels of vimentin were significantly higher in patients with atherosclerotic coronary artery disease than in healthy controls. Western diet-fed mice had more vimentin in serum than normal chow diet-fed mice [12,13].
In the current study, we evaluated how extracellular vimentin affects in adipocyte function by measuring molecular and cellular changes which affect energy intake, storage, and metabolism. While most of studies for vimentin have focused on the role of intracellular vimentin, our current study focuses on the role of extracellular vimentin which elicits signals modulating adipocyte metabolism. Our study aims to reveal a role of extracellular vimentin in the interplay between oxidative stress and adipocyte metabolism.
METHODS
Reagent
Recombinant vimentins (human, mouse) were obtained from American Research Products (Waltham, MA, USA) and from Cloud-clone (Katy, TX, USA). Microcon centrifugal filter devices were from Merck Millipore (Burlington, MA, USA). Insulin, dexamethasone (DEXA), 3-isobutyl-1-methylxanthine (IBMX), thiobarbituric acid, trichloroacetic acid, H-89 dihydrochloride hydrate, and 1,1,3,3-tetramethoxypropane were obtained from Sigma (St. Louis, MO, USA).
Glucose uptake assay kit, FFA uptake assay kit, adenosine triphosphate (ATP) assay kit, L-lactate assay kit, TG assay kit, autophagy assay kit, and deproteinizing sample preparation kit were obtained from Abcam (Cambridge, UK). Mouse vimentin sandwich enzyme-linked immunosorbent assay (ELISA) kit was purchased from LifeSpan BioSciences (Seattle, WA, USA).
Antibodies for GLUT1, GLUT4, phospho-insulin receptor substrate 1 (P-IRS1), P-Akt, phospho-extracellular-signal-regulated kinase (P-ERK), phospho-hormone-sensitive lipase (P-HSL; S563), HSL, peroxisome proliferator-activated receptor γ (PPARγ), P-ERK, inositol-requiring enzyme 1 α (IRE1α), C/EBP homologous protein (CHOP), p62, and microtubule-associated proteins 1A/1B light chain 3 (LC3)-I/II were from Cell Signaling Technology (Danvers, MA, USA). Antibodies for vimentin, GLUT1, insulin-like growth factor 1 receptor (IGF1R), anti-IGF1R antibody, and anti-CD36 antibody were from Abcam. Antibody for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), α-tubulin, and β-actin were from AbFrontier (Seoul, Korea). Antibody for CD36 was from Novus Biologicals (Centennial, CO, USA). Antibody for Caveolin-1 was from BD Biosciences (East Rutherford, NJ, USA).
Cell culture and differentiation
3T3-L1 cells were purchased from American Type Culture Collection (ATCC; CL-173, Manassas, VA, USA) and cultured in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% new born calf serum and 1% penicillin and streptomycin. Once 3T3-L1 cells were confluent, the cells were incubated for additional 48 hours before initiating differentiation. After 2 days post-confluent, the cells were treated with insulin (1 µg/mL), DEXA (1 µM), and IBMX (0.5 mM) in DMEM containing 10% fetal bovine serum (FBS) and 1% P/S. After 2 days, media was replaced with DMEM containing insulin (1 µg/mL), 10% FBS and 1% P/S and kept for 2 days. And the cells were maintained in DMEM containing 10% FBS and 1% P/S until used.
LDL isolation and oxidation
LDL was isolated through density gradient ultracentrifugation. OxLDL was prepared by incubating LDL with 5 µM CuSO₄ in phosphate buffer saline (PBS) for 6 hours at 37°C. To finish the oxidation, LDL was incubated with 100 µM ethylenediaminetetraacetic acid (EDTA) in PBS.
Confirmation of vimentin secretion
Differentiated 3T3-L1 cells were treated with native LDL (50 µg/mL) for 24 hours or oxLDL (50 µg/mL) for 8, 16, 24 hours. The culture medium was harvested from each well, and concentrated using Microcon (Merck Millipore). Vimentin secretion was confirmed using mouse vimentin sandwich ELISA Kit (Lifespan biosciences) and Western blot. Glucose uptake assay
3T3-L1 cells were seeded into a 96-well culture plate at a density of 5,000 cells per well and differentiated. The cells were treated with recombinant vimentin (20 µg/mL) in serum-free media for 24 hours. Thereafter, 3T3-L1-derived adipocytes were tested for glucose uptake using a kit (Abcam). Cells were starved for glucose by pre-incubating with Krebs-Ringer-Phosphate-HEPES (KRPH)/2% bovine serum albumin buffer for 40 minutes and treated with 2-deoxyglucose (2-DG; 10 mM). After 20 minutes, all samples were lysed and 2-deoxyglucose-phosphate (2-DGP) accumulated in the cells was measured using spectrophotometry (absorbance at 412 nm).
Free fatty acid uptake assay
3T3-L1-derived adipocytes were cultured in a 96-well black plate at a density of 80,000 cells per well. After the cells attached to the plate, the cells were treated with vimentin (20 µg/mL) for 24 hours. We added fatty acid dye-loading solution (TF2-C12 Fatty Acid) and measured fluorescence with a fluorescence microplate reader at Ex/Em=485/515 nm.
Autophagy assay
Autophagy activity was measured using the autophagy assay kit (Abcam, #ab139484). 3T3-L1-adipocytes in a 96 well plate were treated with vimentin (20 μg/mL) for 24 hours. The cells were washed with assay buffer, suspended in the dual color detection solution and incubated for 30 minutes at 37°C away from light. Cells were washed twice with assay buffer. Assay buffer (100 µL) was added and fluorescence was read by a microplate reader. The percentage of autophagy was calculated by dividing the signal of autophagosome-staining (480/530 nm) by the signal of nucleus-staining (340/480 nm).
Statistical analysis
Experiments were repeated more than three times and graphs are from collected dataset. To determine if there is a significant difference between the means of two groups, we used Student t-test. Mann-Whitney U test is used when the dependent variable is ordinal, but not normally distributed. The one-way analysis of variance (ANOVA) was used to determine whether there are any statistically significant differences between the means of three or more groups. We used Bonferroni post hoc test to compare pairs in groups. Detailed description of other assays are provided in Supplementary Methods and Supplementary Table 1.
RESULTS
Oxidized LDL via CD36 induces vimentin secretion in adipocytes
We evaluated if adipocytes secrete vimentin in response to oxLDL as macrophages [14]. We treated 3T3-L1-derived adipocytes with or without oxLDL (50 µg/mL). The lipid peroxidation degree of the oxLDL was 8 to 9 nmol malondialdehyde (MDA)/mg of protein in our thiobarbituric acid reactive substance (TBARS) assay. After 24 hours incubation, the culture media were harvested, concentrated and applied onto ELISA. The ELISA showed that oxLDL-induced vimentin secretion in adipocytes (Fig. 1A). We also confirmed oxLDL-induced secretion of vimentin in adipocytes using Western blot. Vimentin secretion was not dependent on oxLDL-incubation time nor oxLDL concentration in a range between 25 and 50 µg/mL (Fig. 1A-C). LDL did not induce vimentin secretion (Fig. 1B-E).
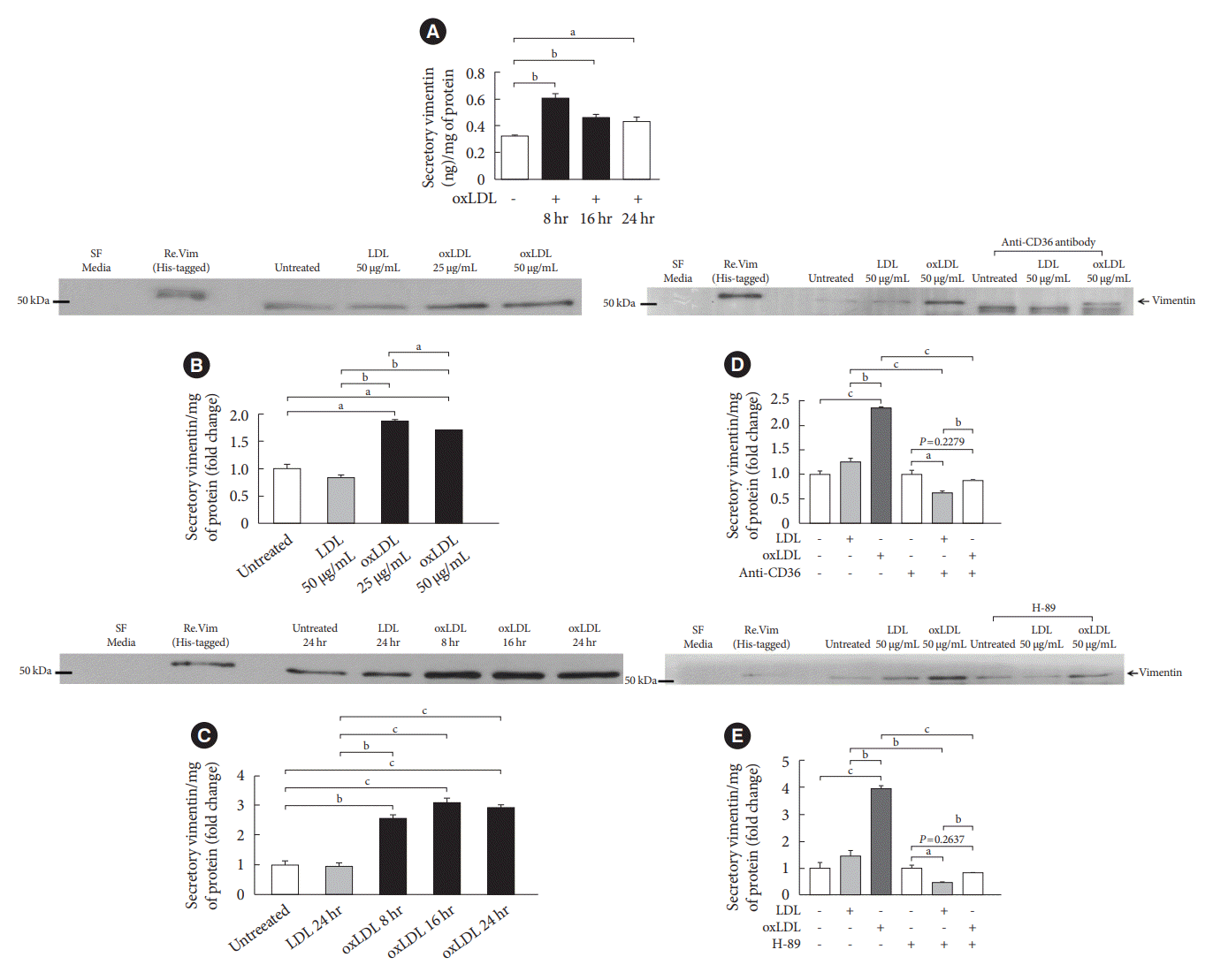
Fig. 1.
Oxidized low-density lipoprotein (oxLDL) induces vimentin secretion in 3T3-L1-derived adipocytes. (A) 3T3-L1-derived adipocytes were treated with or without oxLDL (50 µg/mL) for 8, 16, 24 hours and the concentrations of extracellular vimentin in the media were measured using enzyme-linked immunosorbent assay (ELISA). Data were normalized to the protein amount (mg) of the adipocytes used in the assay (n=3). (B) Detection of vimentin in the concentrated media of 3T3-L1-adipocytes cultured with LDL (50 µg/mL) or different concentrations of oxLDL (25, 50 µg/mL) for 24 hours. In the Western blot, His (molecular weight, 1.6 kDa)-tagged recombinant vimentin was used for a positive control. Data were normalized to the protein amount (mg) of the adipocytes obtained from the individual culture dish (n=3). (C) Detection of vimentin in the concentrated media of 3T3-L1-adipocytes cultured with LDL (50 µg/mL for 24 hours) or oxLDL (50 µg/mL for 8, 16, and 24 hours) using Western blotting. Data were normalized to the cellular protein (mg) (n=3). (D) 3T3-L1-adipocytes were pretreated with anti-CD36 antibody (2 µg/mL) before incubating with LDL (50 µg/mL for 24 hours) or oxLDL (50 µg/mL for 24 hours). Detection of vimentin in the concentrated media of the adipocytes using Western blotting and data were normalized to the cellular protein (mg) (n=3). (E) 3T3-L1-adipocytes were pretreated with or without H-89 (10 µM) before incubating with LDL (50 µg/mL for 24 hours) or oxLDL (50 µg/mL for 24 hours). Detection of vimentin in the concentrated media of adipocytes using Western blotting and data were normalized to the cellular protein (mg) (n=3). All experiments were done more than three times. One-way analysis of variance (ANOVA) with Bonferroni post hoc test was done. SF, serum free. aP<0.05, bP<0.01, cP<0.001.
![]()
It is known that oxLDL binds to scavenger receptor CD36 [14]. We tested if CD36 mediates oxLDL-induced secretion of vimentin. Blocking the interaction between CD36 and oxLDL inhibited vimentin secretion, suggesting that CD36 mediates oxLDL-induced vimentin secretion in the adipocytes (Fig. 1D). It is known that phosphorylation of various residues of vimentin affects vimentin dynamics [15]. In our previous study, we showed that oxLDL phosphorylates intracellular vimentin (Ser72) via protein kinase A (PKA) activation [16]. Therefore, we treated adipocytes with H-89, a pharmacological inhibitor for PKA and tested if oxLDL-induced activation of PKA mediates vimentin secretion. The result showed that inhibition of PKA blocked oxLDL-induced secretion of vimentinin in adipocytes (Fig. 1E). We treated 3T3-L1-derived adipocytes with palmitate, another ligand for CD36. However, palmitate did not induce vimentin secretion in adipocytes (Supplementary Fig. 1).
Extracellular vimentin induces adipocyte hypertrophy with increases in intracellular triglyceride and lipid droplets
We evaluated if extracellular vimentin affects adipocyte metabolism. We treated 3T3-L1-derived adipocytes with recombinant vimentin (20 µg/mL) for 24 hours and stained the cells with Oil-Red O (Fig. 2A). Microscopy revealed that vimentin treatment induced 1.55 fold increase of adipocyte sizes (Fig. 2B) and 1.69 fold increase of intracellular LD sizes (Fig. 2C). The intracellular lipid measured by counting the optical density of the extracted Oil-Red O dye from the cells was increased in the adipocytes treated with vimentin (Fig. 2D). Adipocyte size distribution showed that the frequency of large adipocytes of which diameters are more than 70 µm was significantly increased by vimentin treatment (P<0.001) (Fig. 2E). The frequency of droplets sized more than 16 µm (diameter) was significantly increased by vimentin treatment (P<0.001) (Fig. 2F). Treatment with vimentin (20 µg/mL) for 24 hours induced 1.6 fold increase of intracellular TG in the adipocytes (Fig. 2G).
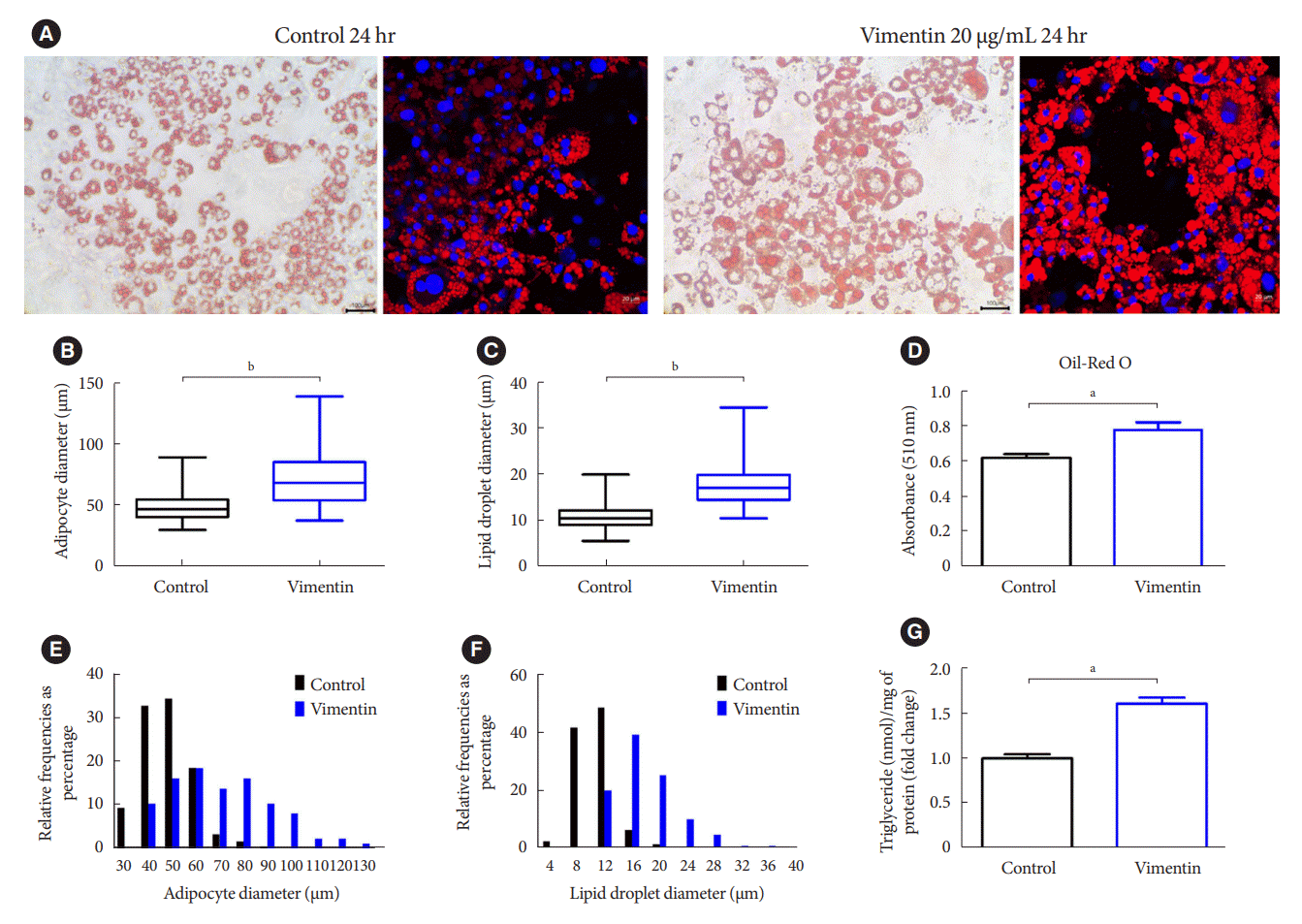
Fig. 2.
Extracellular vimentin induces adipocyte hypertrophy. (A) 3T3-L1-derived adipocytes treated with or without recombinant vimentin (20 µg/mL) for 24 hours, were stained with Oil-Red O and 4ʹ,6-diamidino-2-phenylindole (DAPI). Light microscopy (left panel) and fluorescence microscopy (right panel). Scale bar: 100 µm (left panel), 20 µm (right panel). (B) Comparison of adipocyte diameter (µm) between vimentin-treated (vimentin) and untreated (control) adipocytes. (C) Comparison of lipid droplet diameter (µm) between vimentin-treated (vimentin) and untreated (control) adipocytes. (D) Absorbance of the Oil-Red O dye extracted from the 3T3-L1-derived adipocytes in (A) (n=3). (E) Size distribution of 3T3-L1 adipocytes treated with or without vimentin (20 µg/mL) for 24 hours. (F) Size distribution of intracellular lipid droplets in the 3T3-L1 adipocytes treated with or without vimentin (20 µg/mL) for 24 hours. (G) Intracellular triglyceride was measured using 3T3-L1-derived adipocytes treated with or without recombinant vimentin (20 µg/mL) for 24 hours. Data were normalized to the amount (mg) of total protein from the adipocytes used in the assay (n=3). aP<0.05, bP<0.001.
![]()
Extracellular vimentin promotes glucose uptake and free fatty acid uptake via increasing expression of GLUT1, GLUT4, and CD36 in the plasma membrane of adipocytes
We evaluated if extracellular vimentin affects uptake of glucose and FFA, major energy sources in adipocytes. The 2-DG uptake assay revealed that treatment with vimentin (20 µg/mL) for 24 hours increased glucose uptake by 3.2 fold in the 3T3-L1-derived adipocytes. Insulin as a positive control (1 µM, 30 minutes) increased glucose uptake 3.8 fold in the adipocytes (Fig. 3A). We also measured FFA (dodecanoic acid) uptake in 3T3-L1-derived adipocytes treated with vimentin (20 µg/mL) for 24 hours. The result showed that extracellular vimentin increased FFA uptake 3.2 fold compared to untreated control (Fig. 3B).
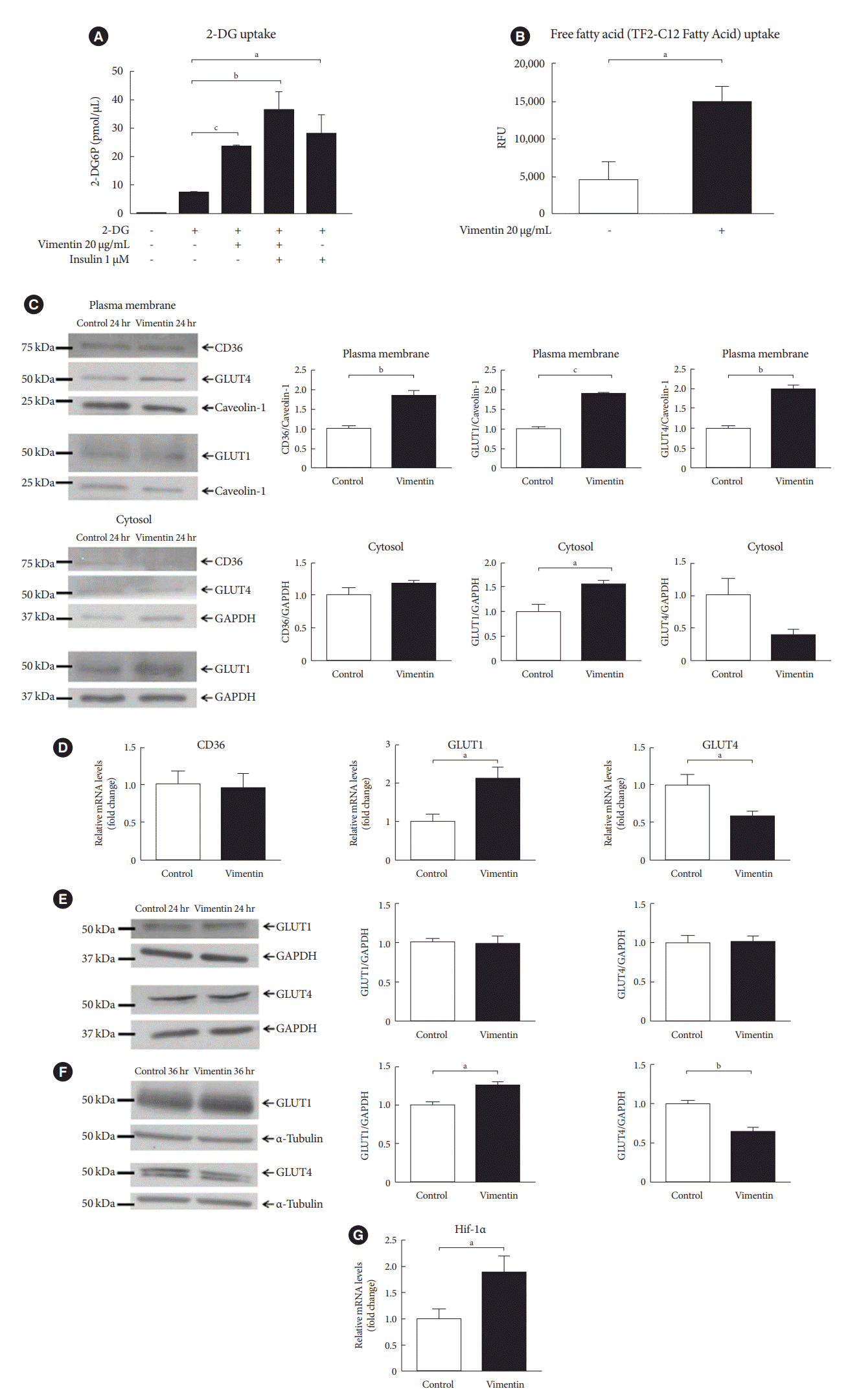
Fig. 3.
Extracellular vimentin promotes glucose uptake and free fatty acid (FFA) uptake via changes in expression of glucose transporters and fatty acid transporter. (A) 3T3-L1-derived adipocytes were treated with recombinant vimentin (20 µg/mL for 24 hours), insulin (1 µM for 30 minutes) or both vimentin (20 µg/mL for 24 hours) and insulin (1 µM for 30 minutes). 2-Deoxyglucose (2-DG) uptake was measured (n=4). (B) 3T3-L1-derived adipocytes were treated with or without recombinant vimentin (20 µg/mL) for 24 hours and FFA uptake was measured using fluorometric fatty acid dye-loading solution (TF2-C12 Fatty Acid) uptake assay (n=3). (C) Western blot analyses for CD36, glucose transporter type 4 (GLUT4), and GLUT1 were performed using fractionated lysates (plasma membrane and cytosol) of 3T3-L1-derived adipocytes treated with or without recombinant vimentin (20 µg/mL) for 24 hours. Band quantification was performed using either caveolin-1 (BD Biosciences) as a plasma membrane marker, or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a cytosol marker (n=3). (D) Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) analyses for CD36, GLUT1, and GLUT4 were performed using RNA from 3T3-L1-derived adipocytes cultured with or without recombinant vimentin (20 µg/mL) for 24 hours. GAPDH was used as an internal control (n=5, n=6, n=4). (E) Western blot analyses for GLUT1 and GLUT4 using total lysates of 3T-3L1-derived adipocytes treated with or with recombinant vimentin (20 µg/mL) for 24 hours. Band quantification was performed using GAPDH, as an internal control (n=3). (F) Western blot analyses for GLUT1 and GLUT4 were performed using total lysates of 3T3-L1-derived adipocytes with or with recombinant vimentin (20 µg/mL) for 36 hours. Band quantification was performed using α-tubulin (AbFrontier), an internal control (n=3). (G) qRT-PCR analysis for hypoxia-inducible factor 1α (Hif-1α) was performed using RNA from 3T3-L1-derived adipocytes treated with or without recombinant vimentin (20 µg/mL) for 24 hours. GAPDH was used as an internal control (n=4). RFU, relative fluorescence unit. aP<0.05, bP<0.01, cP<0.001.
![]()
Based on our findings that extracellular vimentin increases glucose uptake and FFA uptake, we measured glucose transporters including GLUT1, GLUT4, and a fatty acid translocator CD36 in the plasma membrane of adipocytes treated with or without vimentin. Western blots using fractionated cell lysates showed that GLUT1, GLUT4, and CD36 were increased in the plasma membrane 24 hours after the treatment with vimentin (20 µg/mL). Cytosolic expression of GLUT4 and CD36 were not different between vimentin-treated and untreated adipocytes; however, cytosolic expression of GLUT1 was significantly higher in the vimentin-treated adipocytes than in the untreated adipocytes (Fig. 3C). Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) showed that vimentin treatment (20 µg/mL for 24 hours) induced 2.1 fold increase of GLUT1 mRNA while vimentin treatment decreased GLUT4 mRNA by 42%. There was no significant difference in mRNA levels of CD36 between vimentin-treated and untreated adipocytes (Fig. 3D). Based on the data showing that GLUT1 mRNA was increased but GLUT4 mRNA was decreased by vimentin, we measured GLUT1 and GLUT4 proteins in 3T3-L1-derived adipocytes treated with or without vimentin for 24 hours or 36 hours. The result showed that 24-hour-treatment with vimentin did not induce changes in GLUT1 and GLUT4 protein levels in the adipocytes (Fig. 3E). However, vimentin treatment for 36 hours induced 24% increase of GLUT1 protein and 34% decrease of GLUT4 protein in the adipocytes, in accordance with the changes in mRNA levels (Fig. 3F). We also found that hypoxia-inducible factor 1α (Hif-1α), a transcription factor for GLUT1 was upregulated by extracellular vimentin (Fig. 3G).
Extracellular vimentin does not affect phosphorylation of IGF1R, IRS1, Akt, and HSL, but increases transcription of Hif-1α and GLUT1
It has been reported that signaling provoked by insulin via IRS1 promotes GLUT4 trafficking into plasma membrane [17]. In addition, phosphorylation of Akt, a downstream effector of insulin/insulin-like growth factor 1 (IGF1) signaling, promotes expression of GLUT1 in plasma membrane [18]. A recent report showed that extracellular vimentin binds to IGF1R in neurons [19]. Therefore, we examined if extracellular vimentin affects insulin/IGF1 signaling including phosphorylation of IGF1R, IRS1, and Akt. Our Western blot revealed that phospho-IGF1R (Tyr 1158/1162/1163), phospho-IRS1 (Tyr 612), and phospho-Akt (Ser473) were not changed by extracellular vimentin in 3T3-L1-derived adipocytes (Fig. 4A).
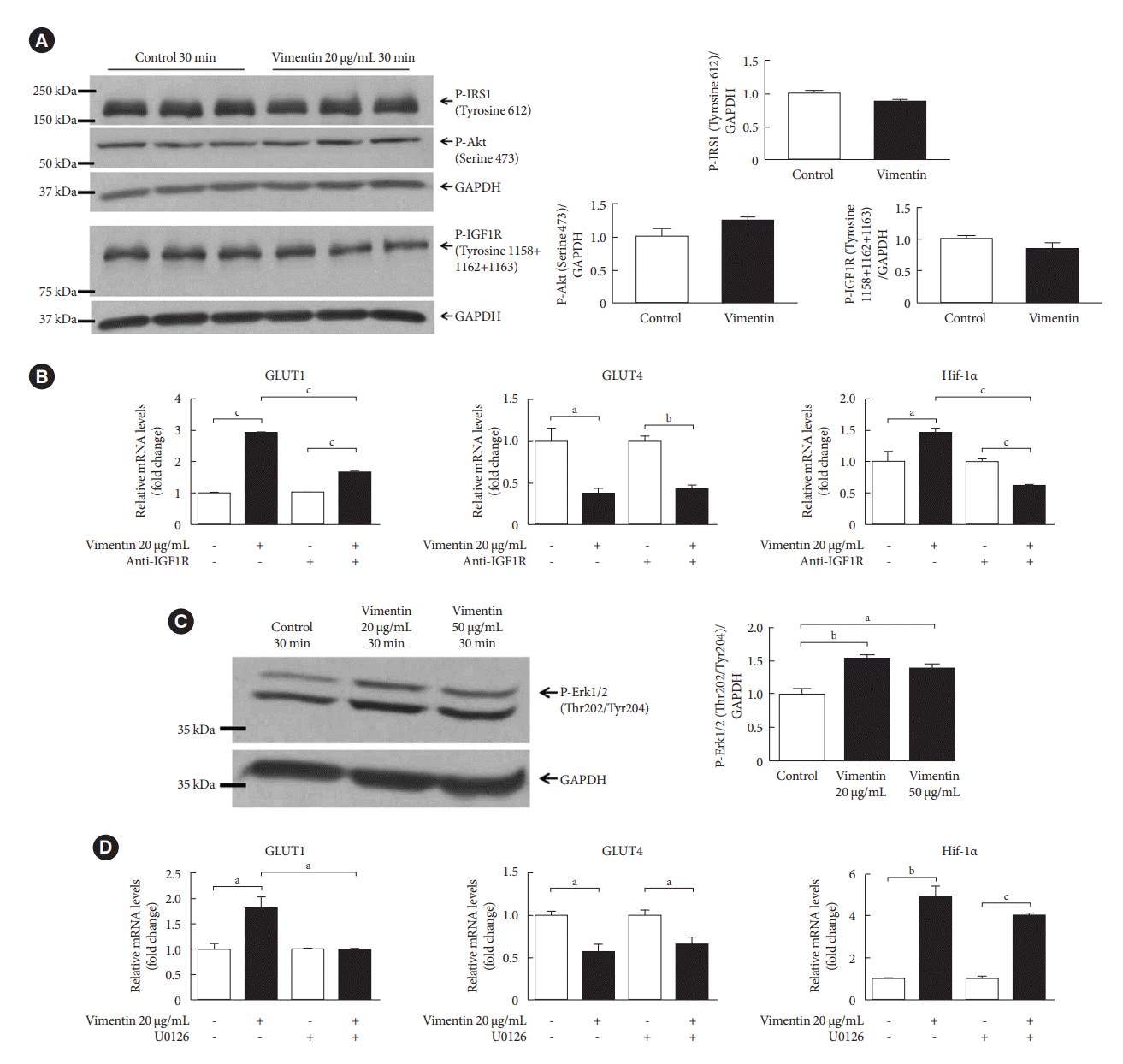
Fig. 4.
Extracellular vimentin-induced glucose transporter type 1 (GLUT1) expression depends on insulin-like growth factor 1 receptor (IGF1R) and activation of extracellular-signal-regulated kinase (ERK). (A) Western blot analyses for phospho-insulin receptor substrate 1 (P-IRS1; Tyrosine 612), P-Akt (Serine 473), and P-IGF1R (Tyrosine 1158+1162+1163) were performed using lysates of 3T3-L1-derived adipocytes treated with or without recombinant vimentin (20 µg/mL) for 30 minutes. Band quantification was performed using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal control. Samples from three different batches of treatment were loaded onto a gel (n=3). (B) Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) analyses for GLUT1, GLUT4, hypoxia-inducible factor 1α (Hif-1α) were performed with RNA from 3T3-L1-derived adipocytes treated or untreated with anti-IGF1R antibody (11 µg/mL) for 1 hour and then incubated with or without recombinant vimentin (20 µg/mL) for 24 hours. GAPDH was used as an internal control (GLUT1, n=3; GLUT4, n=3; Hif-1α, n=4). (C) Western blotting for P-ERK1/2 (Threonine 202/Tyrosine 204) was performed using lysates of 3T3-L1-derived adipocytes treated with or without recombinant vimentin (20 µg/mL) for 30 minutes. Band quantification was performed using GAPDH as an internal control (n=3). (D) qRT-PCR analyses for GLUT1, GLUT4, Hif-1α were performed with RNA from 3T3-L1-derived adipocytes treated or untreated with U0126 (ERK inhibitor, 25 µM) for 1 hour and then incubated with or without recombinant vimentin (20 µg/mL) for 24 hours. GAPDH was used as an internal control (n=3 for each). aP<0.05, bP<0.01, cP<0.001.
![]()
IGF1R has many active phosphorylation sites other than Tyr 1158/1162/1163 [20]. We blocked IGF1R with an antibody and measured mRNA levels of GLUT1, GLUT4, and Hif-1α in the vimentin-treated and untreated adipocytes. Our results showed that blocking IGF1R suppressed the increase in the mRNA levels of GLUT1 and Hif-1α. However, blocking IGF1R did not inhibit the decrease in GLUT4 mRNA (Fig. 4B).
A previous study showed that ERK regulates GLUT1 mRNA expression in cardiomyocytes [21]. Our Western blot revealed that extracellular vimentin increased ERK phosphorylation in 3T3-L1-derived adipocytes (Fig. 4C).
We treated 3T3-L1-adipocytes with U0126 (25 µM), an ERK inhibitor for 1 hour, and then with vimentin for 24 hours. Our qRT-PCR showed that inhibiting ERK blocked the vimentininduced increase of GLUT1. However, ERK inhibition did not affect vimentin-induced decrease of GLUT4 and vimentin-induced Hif-1α expression (Fig. 4D).
Extracellular vimentin has no effect on lactic acid secretion and intracellular ATP level in adipocytes
Since vimentin promotes glucose uptake and expression of Hif-1α and GLUT1, key regulators in glycolysis [22], we performed L-lactate assay and ATP assay to investigate the effect of extracellular vimentin in glycolysis and energy metabolism. The result showed that there was no change in extracellular lactic acid (Fig. 5A) and intracellular ATP level (Fig. 5B) in 3T3-L1-adipocytes treated with vimentin (20 µg/mL) for 24 hours. Overall, our data suggested that energy intake was increased by extracellular vimentin, but energy expenditure was not changed.
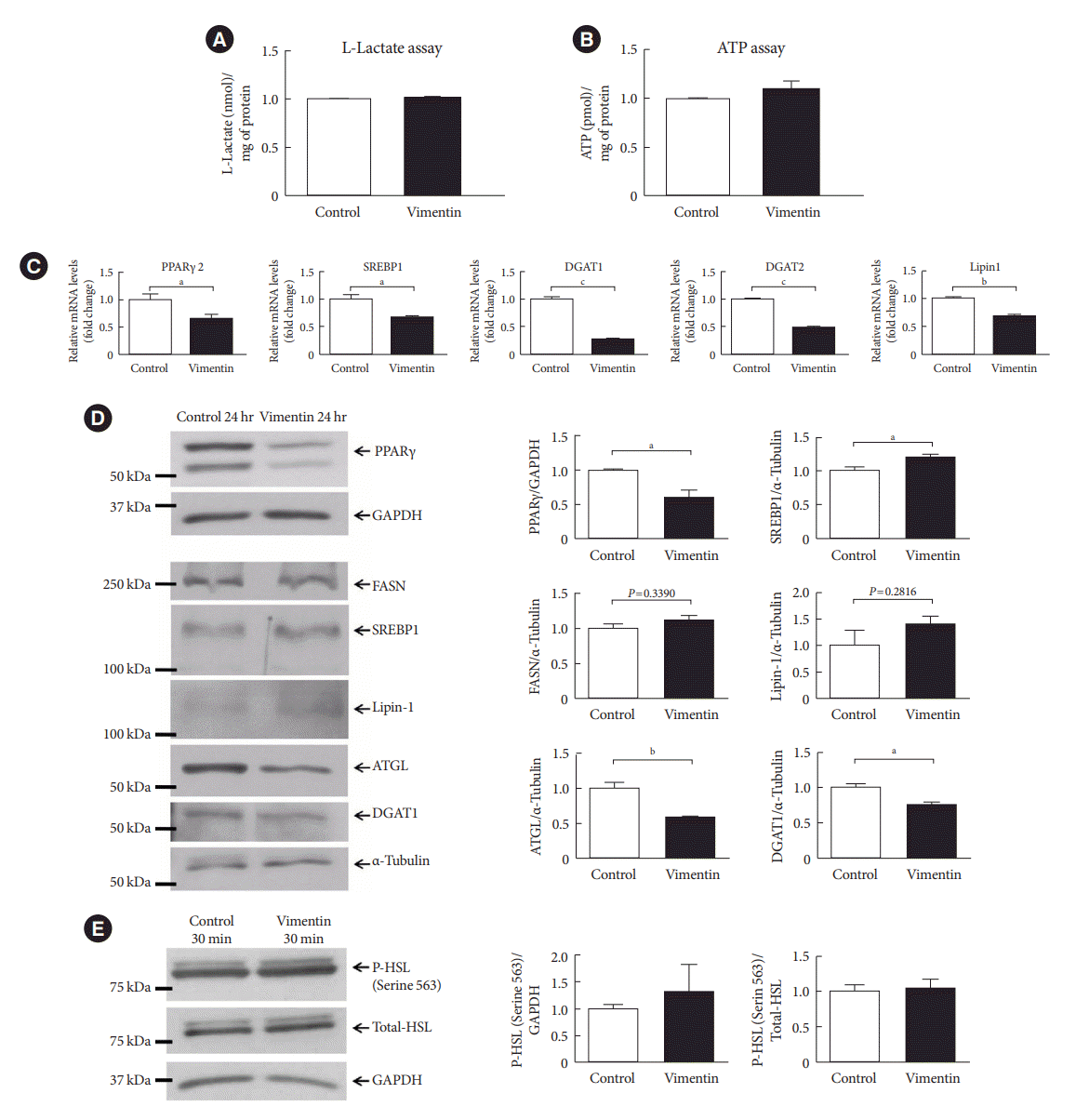
Fig. 5.
Extracellular vimentin modulates expression of molecules for lipogenesis and lipolysis. (A) L-Lactate was measured in the culture media of 3T3-L1-derived adipocytes treated with or without recombinant vimentin (20 µg/mL) for 24 hours. Data were normalized to the protein amount (mg) of the adipocytes used in the assay (n=2). (B) Intracellular adenosine triphosphate (ATP) was measured in 3T3-L1-derived adipocytes treated with or without recombinant vimentin (20 µg/mL) for 24 hours. Data were normalized to the protein amount (mg) of the adipocytes used in the assay (n=4). (C) Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) analyses for peroxisome proliferator-activated receptor γ 2 (PPARγ2), sterol regulatory element-binding protein 1 (SREBP1), diacylglycerol O-acyltransferase 1 (DGAT1), DGAT2, and Lipin1 were performed using RNA from 3T3-L1-derived adipocytes treated with or without recombinant vimentin (20 µg/mL) for 24 hours. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control (n=4, n=3, n=3, n=3). (D) Western blot analysis for PPARγ, fatty acid synthase (FASN), SREBP1, Lipin1, adipose triglyceride lipase (ATGL), and DGAT1 were performed using lysates of 3T3-L1-derived adipocytes treated with or without recombinant vimentin (20 µg/mL) for 24 hours. Band quantification was performed using GAPDH and α-tubulin as internal controls (n=3). (E) Western blot analysis for phosphor-hormone-sensitive lipase (P-HSL; Serine 564) was performed using lysates of 3T3-L1-derived adipocytes treated with or without recombinant vimentin (20 µg/mL) for 30 minutes. Band quantification was performed using total HSL and GAPDH as internal controls (n=3). aP<0.05, bP<0.01, cP<0.001.
![]()
Extracellular vimentin reduces expression of molecules involved in lipogenesis and lipolysis
We also evaluated factors associated with lipogenesis and lipolysis. Our data showed that transcription of factors involved in lipogenesis such as PPARγ, sterol regulatory element-binding protein 1 (SREBP1), diacylglycerol O-acyltransferase 1 (DGAT1) and 2, and Lipin1 were significantly decreased in adipocytes treated with vimentin for 24 hours (Fig. 5C). Western blot also revealed that PPARγ and DGAT1 were decreased in the vimentin-treated adipocytes. However, protein levels of fatty acid synthase (FASN) and Lipin1, a phosphatidate phosphatase (PAP) which catalyzes phosphatidic acid to diacylglycerol [23] were not different between vimentin-treated and untreated adipocytes (Fig. 5D).
Adipose triglyceride lipase (ATGL) is known to catalyze the initial step in adipose TG lipolysis, working in concert with other enzymes to mobilize TG for energy production [24]. Our Western blot showed that ATGL was decreased in the vimentin-treated adipocytes (Fig. 5D). In addition, extracellular vimentin did not affect phosphorylation of HSL (Fig. 5E).
Extracellular vimentin induces endoplasmic reticulum stress and impairment of autophagy
It has been reported that hypertrophied adipocytes are exposed to cellular stresses as hypoxia [8]. Hypoxia is known to induce endoplasmic reticulum (ER) stress, an aggravating factor for obesity [25,26]. Therefore, we evaluated if extracellular vimentin induces ER stress in adipocytes. Western blot showed that extracellular vimentin increased expression of ER stress markers including protein kinase R-like ER kinase (PERK), IRE1α, and CHOP by 1.27, 1.72, and 2.78 folds, respectively (Fig. 6A). In addition, qRT-PCR also revealed that CHOP and binding immunoglobulin protein (BiP) mRNAs were significantly increased in vimentin-treated adipocytes (Fig. 6B).
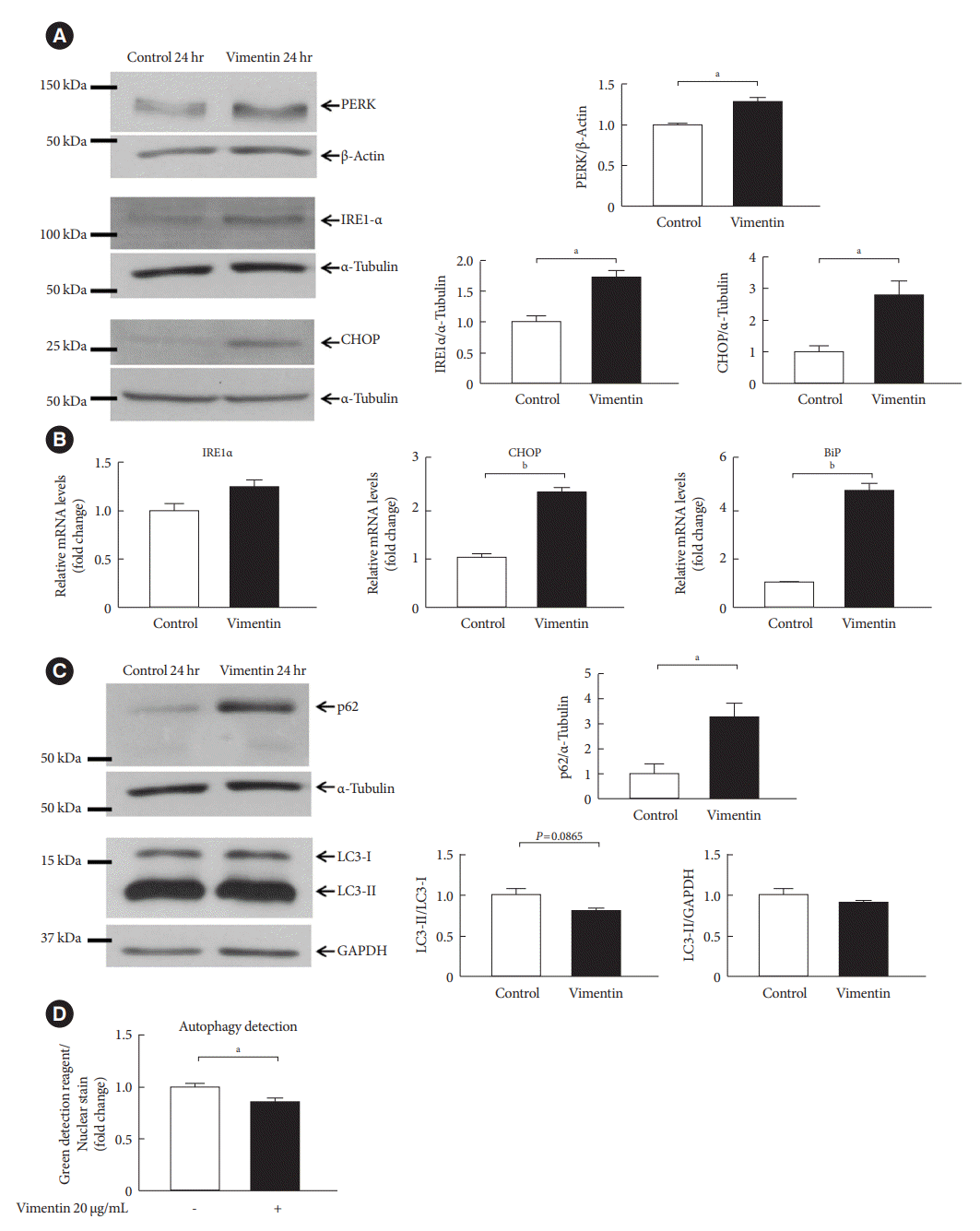
Fig. 6.
Extracellular vimentin induces endoplasmic reticulum (ER) stress and impairs autophagy. (A) Western blot analyses for protein kinase R-like ER kinase (PERK), inositol-requiring enzyme 1 α (IRE1α), and C/EBP homologous protein (CHOP) were performed using lysates of 3T3-L1-derived adipocytes treated with or without recombinant vimentin (20 µg/mL) for 24 hours. Band quantification was performed using β-actin and α-tubulin as internal controls (n=3). (B) Quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) analyses for IRE1α, CHOP, and binding immunoglobulin protein (BiP) were performed using RNA from 3T3-L1-derived adipocytes treated with or without vimentin (20 µg/mL) for 24 hours. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control (n=4). (C) Western blot analysis for p62, light chain 3 (LC3)-I, and LC3-II were performed using lysates of 3T3-L1-derived adipocytes treated with or without recombinant vimentin (20 µg/mL) for 24 hours. Band quantification was performed using α-tubulin and GAPDH as internal controls (n=3). (D) Autophagic vacuoles in 3T3-L1-derived adipocytes treated with or without recombinant vimentin (20 µg/mL) for 24 hours, were measured. Data were normalized to Hoechst 33342 staining in the adipocytes used in the assay (n=5). aP<0.05, bP<0.001.
![]()
There have been reports revealing that ER stress and autophagy are dynamically interconnected, and obesity and its associated stress often interfere with autophagy [27]. Our Western blots revealed that extracellular vimentin-induced accumulation of p62, which is a key feature of impaired autophagy (Fig. 6C). Vimentin-induced autophagy impairment was also detected by autophagy assay which quantitates fluorescent staining of autophagolysosomes (Fig. 6D).
We concluded that extracellular vimentin induces adipocyte hypertrophy with increased energy storage via increasing transcription of GLUT1 and Hif-1α, promoting trafficking of GLUT4 and CD36, and thus increasing glucose and fatty acid uptake. Extracellular vimentin also induces ER stress, impairs autophagy, regulates expression of molecules related to lipogenesis and lipolysis and thus modulates energy metabolism in adipocytes.
DISCUSSION
Vimentin filament network plays an important role in maintaining cellular architecture and intracellular transport of molecules [28]. Vimentin has been studied for its entity of structural protein. Nevertheless, role of vimentin as a signaling molecule has not been studied extensively. In our previous study, we revealed that vimentin contributes to development of obesity and diabetes, but the mechanism was not elucidated. Vimentin null mouse fed a high fat diet showed less adiposity and improved glucose tolerance and adipocytes from vimentin null mouse showed less plasma membrane expression of CD36 and GLUT4 [10]. Although we reported that vimentin is secreted extracellularly in an inflammatory milieu with oxidative stress [12], effects of extracellular vimentin in adipocyte metabolism has not been studied. OxLDL, a marker of oxidative stress is known to activate inflammatory processes in a variety of cells including macrophages [29]. OxLDL also promotes hypoxia-induced expression of adipocytokines in adipocytes and contributes to adipose tissue inflammation. OxLDL and adipose tissue inflammation impair insulin-mediated glucose uptake in adipocytes [30].
In the current study, we show that minimally oxLDL (7 to 9 nmol MDA/mg of LDL protein) via CD36 induces vimentin secretion in adipocytes. In our experiments, 3T3-L1-derived adipocytes treated with vimentin for 24 hours showed increased membrane expression of GLUT1, GLUT4, and CD36. GLUT1 is known to be insulin-independent and broadly distributed in different tissues [31]. GLUT1 is induced by cellular stresses as hyperosmolarity [32]. In contrast, GLUT4 is insulin-dependent and is the major glucose transporter in adipocytes [17]. In adipocytes, active Akt functions in translocation of GLUT4 and GLUT1 to the plasma membrane [33]. CD36 expression is enhanced by oxLDL [34]. We previously reported that intracellular vimentin phosphorylation induced by oxLDL promotes CD36 trafficking to the plasma membrane [18]. In the current study, extracellular vimentin-induced trafficking of GLUT1, GLUT4, and CD36 are independent of insulin/IGF1 signaling including phosphorylation of Akt (Fig. 4A). Increased membrane expression of GLUT1, GLUT4, and CD36 results in increased uptake of glucose and fatty acid (Fig. 3). On transcriptional levels, extracellular vimentin increases GLUT1 but decreases GLUT4 (Fig. 3) and this is similar to differential effects of inflammatory cytokines on expression of both GLUTs [35,36]. Cytokines downregulate GLUT4, upregulate GLUT1 and stimulate hexose transport in fibroblasts or in rat adipocytes [37,38]. This process is known to contribute to development of insulin resistance. Obese and T2DM patients show reduced response to insulin and thus less expression of hexokinase II which phosphorylates GLUT4 [39]. Enhanced glucose transport through GLUT1 and availability for the formation of hexosamines through hexokinase I are the common feature for the insulin resistance induced by obesity, inflammation and infection. Decreased GLUT4 expression and decrease in hexokinase II activity lead to a decrease in insulin-mediated glucose transport. Therefore, our finding suggests vimentin secreted by oxLDL may contribute to insulin resistance.
In adipocytes treated with vimentin, increased energy sources seemed to be stored as TG rather than being utilized to generate ATP. TGs are stored in cytosolic LDs of adipocytes. TGs regulate energy balance through lipolysis. If the energy demand increases, hydrolytic cleavage of TG produces non-esterified fatty acids (NEFA) which can be used for generating energy [40]. However, elevated NEFA in serum promotes metabolic derangement such as insulin resistance [41]. We found that extracellular vimentin increases uptake and storage of energy as a form of TG. Extracelluar vimentin deteriorates energy balance and causes an anabolic effect in adipocytes. Adipose tissue acts as an endocrine organ modulating energy homeostasis. There are many “adipokines” secreted by adipocytes regulating energy homeostasis [8]. In this regard, role of extracellular vimentin appears to be similar to a function of an adipokine.
In our experiment, extracellular vimentin induces enlargement of adipocytes and increases sizes of LDs. Hypertrophic adipocytes cause a decrease in oxygen consumption and oxidant production in mitochondria [42]. In accordance, our experiment revealed that extracellular vimentin increases transcription of Hif-1α. It has been reported that Hif-1α is induced by hypoxia and mediates a decrease of oxygen consumption. Obesity causes an increase in Hif-1α mRNA and protein in adipose tissue [43]. Hif-1α induces hypoxic gene expression and inhibits TCA cycle by activating pyruvate dehydrogenase kinase 1 (PDK1) [44]. A previous study showed that deletion of Hif-1α increases ATP levels and decreases ROS generation [44]. Another study reported that in hypoxic condition, cells reduce oxidative metabolism, increase glucose uptake and thus maintain cellular energy [45]. Extracellular vimentin increases mRNA level of Hif-1α, a known transcription factor for GLUT1 [46] and thus increases GLUT1 expression (Fig. 5).
Moreover, our experiment showed that extracellular vimentin increases ER stress and decreases autophagy (Fig. 6). It has been reported that hypertrophied adipocytes are exposed to cellular stresses as hypoxia [10] and hypoxia is known to induce ER stress, an aggravating factor for obesity [26]. Adipose tissue dysfunction is caused by excess energy in over-nutrition. Continuous need to store TG induces adipocyte hypertrophy with synthesis of more proteins for LD formation, and causes ER stress. Obesity-induced ER stress causes adipose tissue inflammation (lipotoxicity) [9]. ER stress alters expression of genes involved in maintaining energy and lipid homeostasis. ER stress down-regulates genes for lipogenesis [47]. In our study, extracellular vimentin causes ER stress and down-regulates genes for lipogenesis including PPARγ, SREBP1, DGAT1 and 2 (Fig. 5C). Lipin1 is an enzyme for lipid synthesis and as a nuclear receptor coactivator playing important roles in lipid homeostasis and metabolism [23]. A previous research showed that Lipin1 transcription was suppressed by ER stress and activation of PPARγ recovered the ER stress-induced Lipin1 suppression [48]. In our data, Lipin1 transcription was down-regulated by vimentin, but Lipin1 protein level was not affected by vimentin (Fig. 5C and D). Therefore, it seemed that ER stress and PPARγ decrease induced by vimentin, suppressed Lipin1 transcription despite increase of Hif-1α, a transcription factor for Lipin1.
We found that extracellular vimentin decreases autophagy. Recent investigations have revealed that ER stress and autophagy are dynamically interconnected and that ER stress can either stimulate or inhibit autophagy [27]. Vimentin-induced autophagy impairment and decrease of lipases as ATGL may facilitate adipocyte hypertrophy.
Obesity develops with adipose tissue expansion characterized by adipocyte hypertrophy which is a known stressor for adipocytes, and adipose tissue inflammation which contributes to eliciting signals for insulin resistance in adipocytes [1]. When lipid accumulation exceeds maximum storage capacity, adipocytes increase lipolysis and release fatty acids into circulation [7]. Subsequently, ectopic fat accumulates in peripheral organs and the adipocyte dysfunction eventually leads to abnormal glucose metabolism and systemic insulin resistance. This phenomenon has been commonly referred to as “lipotoxicity.” Therefore, adipocyte hypertrophy is a hallmark of obesity, suggesting a potential for metabolic dysfunction. A recent research of Baldini et al. [42] showed that hypertrophied adipocytes show increased cytosolic TG, large LDs, impaired mitochondrial oxygen consumption, and reduced oxidative phosphorylation protein levels compared to mature adipocytes in their in vitro early adipocyte hypertrophy model. In parallel, our data showed that extracellular vimentin induces adipocyte hypertrophy with key features of dysregulation of energy homeostasis with an increase in ER stress and a decrease in autophagy. Vimentin is released by oxLDL and thus may be abundant in inflammatory adipose tissue. Therefore, our study may suggest a mechanism of how metabolic inflammation promotes adipocyte hypertrophy and dysfunctional metabolism. Extracellular vimentin may link these pathologic processes. OxLDL induces vimentin secretion in adipocytes and extracellar vimentin affects adipocyte metabolism in an autocrine and a paracrine manners. Extracellular vimentin secreted by macrophages may also affect adipocyte metabolism in inflamed adipose tissue. Therefore, vimentin may mediate intercellular communication between macrophages and adipocytes. Many clinical and basic studies have shown that obesity leads to inflammation in various tissues and conversely, leads subjects to be more susceptible to obesity and insulin resistance [1]. Mechanisms by which obesity promotes inflammation have been suggested and proinflammatory role of oxidized lipids is a part. However, mechanism of how inflammation with oxidative stress leads to obesity has not been extensively studied.
In our current research, we suggest vimentin as a molecule functioning in the crosstalk between inflammation and obesity. Extracellular vimentin secreted by oxLDL alters energy metabolism in adipocytes and promotes adipocyte hypertrophy, leading to development of obesity.
 XML Download
XML Download