

 PDF
PDF Citation
Citation Print
Print



Introduction
Intracranial arterial disease (ICAD) is a common cause of ischemic stroke worldwide, particularly in Asian and African populations [1]. However, treatment options for ICAD are more limited than those for extracranial carotid atherosclerosis. ICAD tends to respond poorly to medical treatment, resulting in a higher risk of recurrent ischemic stroke [2]. Furthermore, despite recent advancements in endovascular techniques, permanent stenting of the intracranial artery is still regarded as having more risk than benefit [3].
In contrast to extracranial carotid disease, ICAD is not fully characterized because of the small size of the intracranial arteries and their deep location in the skull. In addition, the tortuosity of the intracranial carotid artery contributes to challenges associated with in vivo diagnostic imaging tools and effective treatments [4]. Furthermore, because it is difficult to obtain tissue samples from patients with ICAD, pathology–imaging correlation studies are difficult to conduct. Therefore, it is difficult to study ICAD actively.
Nevertheless, significant advances have been made in the understanding of the pathogenesis and diagnostic approaches for ICAD over the past decade. It has been argued that ICAD is a heterogeneous disease that includes diseases other than atherosclerosis [5]. In addition, with advances in magnetic resonance (MR) technology, high-resolution vessel wall imaging (HR-VWI) has been used in clinical practice to visualize the vascular walls of intracranial arteries with luminal diameter of less than 2–4 mm [6]. These two conceptual and radiological advantages have prompted a shift in our approach to ICAD in clinical practice and its management.
This review will first summarize how the intracranial artery differs from the extracranial artery in terms of anatomy, embryology, and histology. Based on these fundamental differences, we aimed to elucidate the heterogeneous mechanisms and pathophysiology of ICAD. Here, we review how these different ICAD etiologies can be visualized and distinguished using HR-VWI. Finally, we explore the clinical applications of HR-VWI and potential individualized approaches, with an emphasis on the temporal changes observed on HR-VWI.
Go to : 
Search strategy and study selection criteria
PubMed was searched using the search terms “intracranial atherosclerosis,” “intracranial stenosis,” and “high-resolution vessel wall imaging” to identify relevant studies published until July 2023. Articles published in English were reviewed. References from relevant articles and reviews were also identified. The final reference list was generated based on originality and relevance to the topic.
Go to :
Terminology
We used the term intracranial arterial disease (ICAD) to describe the category of intracranial arterial diseases that can lead to vessel constriction [6,7]. ICAD includes diseases with specific vasculopathies that may lead to morphological features of vessel constriction. ICAD includes atherosclerosis, moyamoya disease (MMD), vasculitis, and dissection. The term indicates that the etiology of vascular narrowing can be heterogeneous.
Go to :
Intracranial arteries compared to extracranial arteries
Anatomy
The anatomical differences between the extracranial and intracranial arteries are as follows. The intracranial cerebral arteries are relatively small in diameter and travel in the subarachnoid space, floating in the cerebrospinal fluid (CSF). Intracranial arteries have many perforating branches that run perpendicularly, are more tortuous than extracranial arteries, and have abundant collateral circulation (Figure 1A). In contrast, extracranial arteries are relatively large in diameter, surrounded by connective tissue, lack perforating arteries, and are less tortuous.
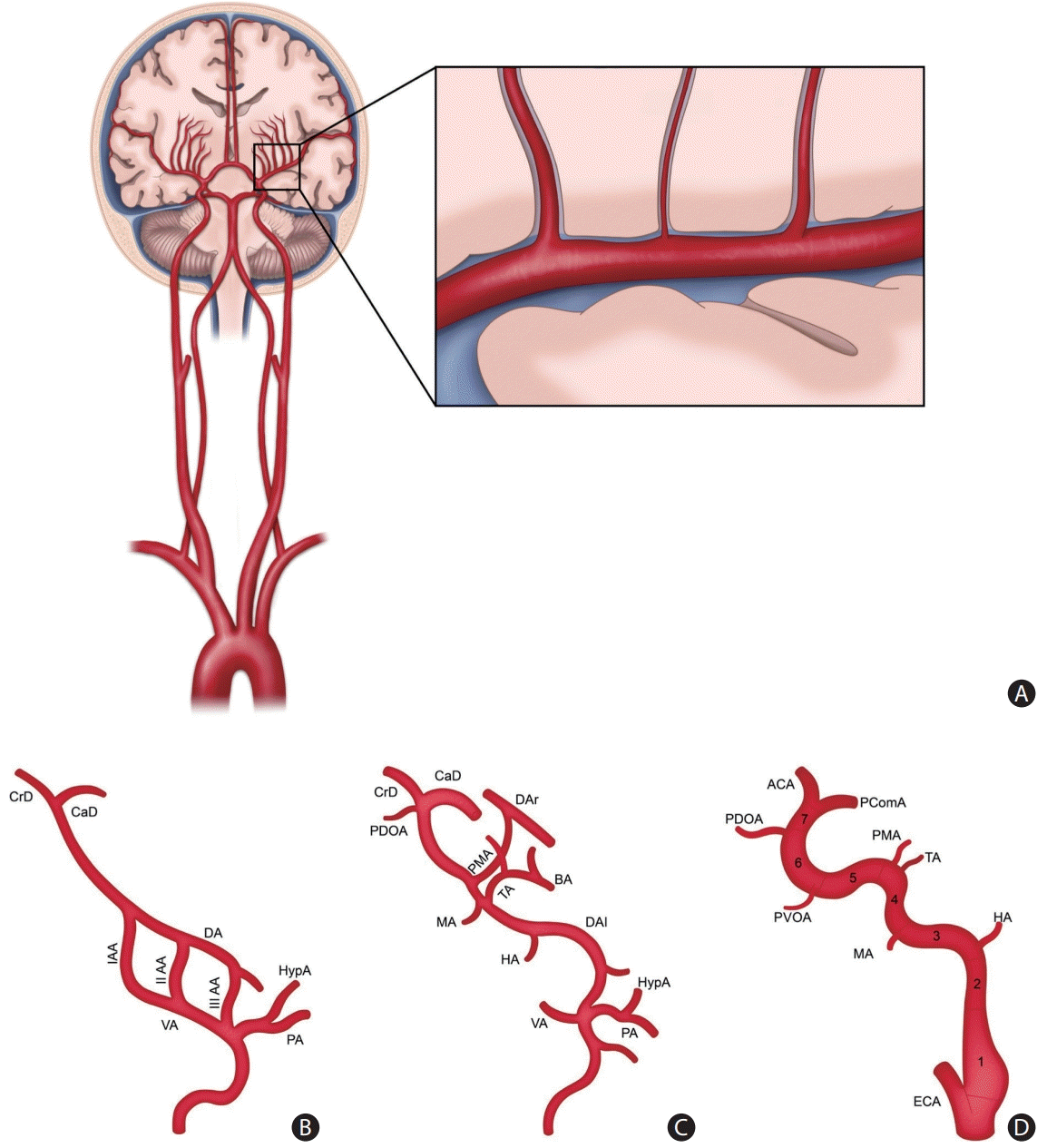 | Figure 1.Overview of cerebrovascular anatomy and embryology. (A) Schematic drawing of the anatomy of the intracranial and extracranial arteries. The middle cerebral artery traverses through the subarachnoid space and extends perforators into the cerebral parenchyma. (B) In the early embryo, the three aortic arches (AA) link the ventral (VA) and dorsal aorta (DA). (C) As the first (I AA) and second aortic arches (II AA) regress, the DA and the third aortic arch (III AA) remain as the primitive internal carotid artery (ICA). The remnant I AA corresponds to the mandibular artery (MA), and the remnant II AA corresponds to the hyoid artery (HA). Other embryonic arteries arise, such as the primitive maxillary artery (PMA), trigeminal artery (TA), and primitive dorsal ophthalmic artery (PDOA). (D) The embryonic arteries that regress divide the primitive ICA into seven segments. The first segment corresponds to the remnant of the III AA. The second segment is the remnant of the DA between the II AA and III AA. The third to seventh segments are divided into the MA, PMA, PDOA, primitive ventral OA (PVOA), and ICA bifurcation. CrD, cranial division; CaD, caudal division; HypA, hypoglossal artery; PA, proatlantal artery; DAr, dorsal aorta right; DAl, dorsal aorta left; BA, basilar artery; ACA, anterior cerebral artery; PComA, posterior communicating artery; ECA, external carotid artery.
|
Embryology
As shown in Figure 1B-D, primitive intracranial arteries arise, regress, and mature during development. Lasjaunias [8] proposed dividing the internal carotid artery (ICA) into seven segments, each with distinct vulnerabilities to pathological changes based on segmental identity and evolutionary/embryological characteristics. There are examples of vascular diseases, such as serpentine arterial aneurysms, arterial dysplasia, dolichoarterial segments, and MMD, that involve only certain ICA segments [8].
In the posterior circulation, the proximal vertebral artery (VA) originates from seventh dorsal cervical segmental arteries, while the rostral portion originates from the bilateral longitudinal neural arteries [9]. The boundary between these two regions is near the site where the extracranial VA transitions to the intracranial vertebrobasilar system [10].
In addition, the endothelial cells of the intracranial arteries are of neural crest origin, whereas those of the extracranial ICA and the vertebrobasilar system, up to the superior cerebellar artery, are of mesodermal origin [11]. In intracranial arterial dolichoectasia, only the ICA, posterior communicating artery, and posterior cerebral artery, which originate from the neural crest, are affected, whereas the rest of the posterior circulation is spared, indicating that the disease is a neurocristopathy [11,12]. Thus, the intracranial arteries in each segment differ on an embryological basis, beyond mere regional or anatomic differences, which provides insight into the types of vascular pathologies that commonly occur in each segment.
Histology
Blood vessels consist of three layers: the inner intimal layer lined with endothelial cells, the middle tunica media containing smooth muscle cells, and the outer tunica adventitia (Figure 2A). The extracranial arteries, including the aorta and carotid arteries, are elastic with abundant elastin filaments in their thick tunica media and adventitia. The adventitial layer is continuous with the surrounding connective tissue. The vasa vasorum (VV) is abundant and supplies oxygen and nutrients to the blood vessels. In contrast, the intracranial artery is a muscular artery with relatively fewer elastic fibers [13]. The intracranial artery “floats” in the subarachnoid space, so the tunica adventitia cannot be directly extended to the connective tissue [14]. Therefore, the media and adventitial layers are relatively thin, and the VV is not extensively developed [14].
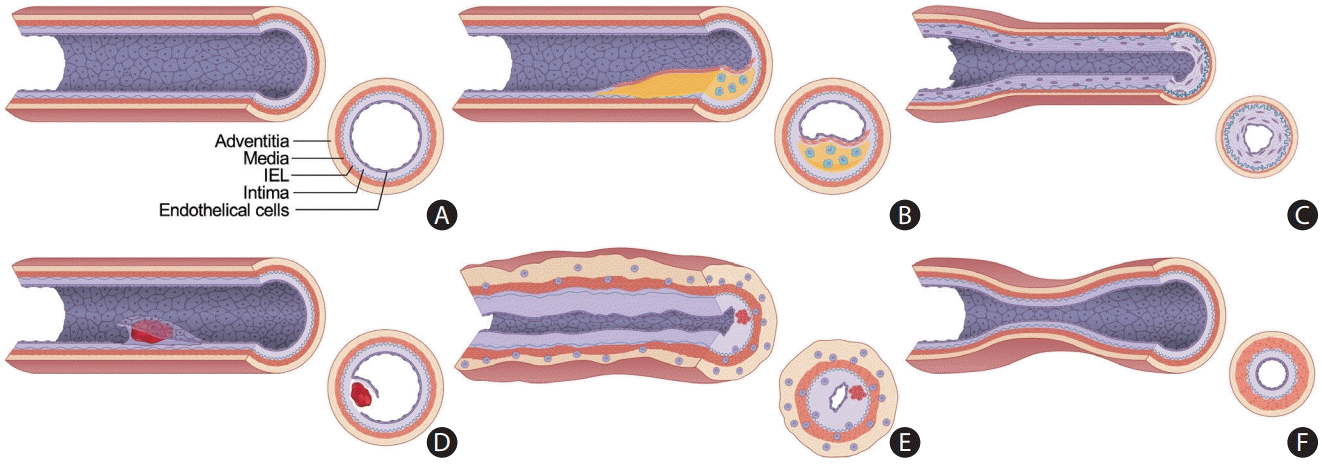 | Figure 2.Schematic diagram displaying the heterogeneity of intracranial arterial diseases. (A) The structure of a normal intracranial vessel wall consists of endothelial cells, intima, internal elastic lamina (IEL), media, and adventitia. There is no external elastic lamina in intracranial arteries. Intracranial arteries have fewer elastin fibers in the media, thinner adventitia, and thicker IEL compared to extracranial arteries. (B) Atherosclerosis is characterized by a lipid core covered by fibrous caps. Infiltrating macrophages are found within the lipid core. More advanced forms of atherosclerosis are infrequently seen in intracranial arteries. (C) Intracranial arteries in moyamoya disease undergo negative remodeling due to fibrosis and thickening of the intima and proliferation of smooth muscle cells. There is also infolding and chronic contraction of the IEL. (D) Intracranial arterial dissection. The intima is torn inward to form an intimal flap and a double lumen. Intramural hematoma is sometimes seen. (E) Mononuclear and granulomatous adventitial inflammation and focal fibrin thrombus formation in vasculitis. Concentric thickening of the vessel wall is seen. (F) In reversible cerebral vasoconstriction syndrome, the diameter of the entire vessel wall is reduced due to the contraction of smooth muscle in the media layer.
|
Histological differences between the intracranial and extracranial arteries likely stem from the different embryological origins of each arterial segment [13]. It has been proposed that the thick adventitia surrounding the extracranial cerebral arteries undergo significant transition as they travel through the skull and pierce the dura. Their histological characteristics gradually transition to those of the intracranial arteries as they travel through the intracranial subarachnoid space [10].
Go to :
ICAD: heterogeneity in their pathogeneses
ICAD: atherosclerosis
Atherosclerotic plaques and the resulting arterial stenosis of the intracranial arteries show histological differences from those of the extracranial cerebral arteries. Intracranial atherosclerosis more often forms a relatively fibrous plaque, while the formation of a lipid-rich necrotic core, often observed in extracranial atherosclerotic plaques, is less frequently observed or restricted to the most proximal intracranial segments [13,15]. In addition, intracranial cerebral arteries after ICA bifurcation do not frequently have calcifications except at the carotid siphons or proximal intracranial VAs (Figure 2B). Interestingly, intracranial atherosclerosis develops and progresses at a relatively later age than that in the coronary and extracranial carotid arteries [15]. In the systemic arterial vasculature, a fatty streak is detected in the aorta during the second decade, and atherosclerosis begins in the carotid arteries during the third decade.
The reasons for these differences in the atherogenic processes in the intracranial and extracranial cerebral arteries are not clearly understood. Several biological and histological differences are believed to play a role in this process. First, the intracranial artery “floats” in the subarachnoid space. The formation of the VV is weak, and it is possible that oxygen and nutrients are supplied via direct diffusion from CSF [14]. Consequently, neovascularization of the VV, which may have a role in the maturation of atherosclerotic plaques, is limited. Intracranial VV does not exist at birth but may develop later in adulthood; however, extracranial vascular beds, including proximal ICA and VA, contain VV-ish structures from birth [16]. Intracranial VV further develops in related to vessel wall thickening, decreased blood flow, and decreased oxygen utilization, which occur in older individuals and those with hypertension [17]. In one study, intracranial atherosclerosis was associated with age, hypertension, and diabetes, whereas hyperlipidemia and smoking were not [18]. It could be interpreted that hypertension and age could promote neovascularization of the VV in the adventitia, whereas hyperlipidemia and smoking do not directly impact the VV because they damage the intima at the initial stage [16]. Second, intracranial arteries have an embryological origin that is distinct from that of extracranial arteries and appear to be relatively insensitive to sympathomimetic stimulation [10]. Third, the shear stress on the intracranial arteries may be partially ameliorated by autoregulation in the distal cerebral vascular beds. Lastly, molecular differences, such as augmented expression of matrix metalloproteinase-9 and fewer caveolae, may explain the differences in the atherogenic process [19].
ICAD: RNF213 spectrum disorder or MMD spectrum
MMD is an uncommon inherited condition that causes progressive steno-occlusion of the distal ICA during early adulthood. It has a relatively high prevalence in East Asia (1.61–18.1/100,000 individuals) (Figure 2C) [20,21].
According to the diagnostic criteria proposed in 2021, MMD can be diagnosed by MR or cerebral angiography when significant steno-occlusion of the terminal ICA with a rich abnormal vascular network in the basal ganglia or perivascular white matter is documented, and other potential causes are excluded, such as autoimmune disease, meningitis, brain tumor, neurofibromatosis, or radiation vasculopathy [22].
Variants in several susceptibility genes are involved in MMD development. Among them, RNF213 14429G>A (rs112735431, resulting in Arg4810Lys) is a major founder mutation in East Asian countries and is found in approximately 80%–90% of families with familial traits of MMD [23]. However, the RNF213 14429G>A variant has a prevalence of approximately 1%–1.5% in the general population in Korea and Japan [24]. Patients with this RNF213 variant do not always present with the typical MMD-like distal ICA occlusions or abundant collaterals. Instead, patients with this variant often present normal intracranial arteries or asymptomatic narrowing of the long segment of the intracranial artery without collateral channels [25]. Although RNF213-related intracranial atherosclerosis is more likely to be associated with negative remodeling and vulnerable to hemodynamic compromise [25], it is difficult to distinguish this condition from atherosclerotic stenosis, which is typically found in middle-aged individuals.
Bang et al. [26] proposed the concept of the RNF213 spectrum disorder. In this disorder, aging, susceptibility genes such as RNF213, and environmental factors interact to develop and progress to ICAD. Their hypothesis suggested that ICAD is likely a result of a mixed pathology; consequently, the progression of ICAD may vary between individuals. The unifying suggestion of RNF213 spectrum disorder is a practical way to evaluate pre-symptomatic ICAD cases and comprehend the pathogenesis of ICAD, particularly for patients of East Asian origin, given the relatively high percentage of carriers of RNF213 variants in the population [26]. However, no pathological studies have been reported, and further research is needed to test the validity of this concept. Additionally, a minority of patients with ICAD have typical MMD-like vascular pathology without an RNF213 mutation [27], and RNF213 polymorphism is reported to be found in intracranial dissection cases and multiple intracranial stenosis [28,29]. More research correlating genetic, pathological, and serial imaging data is needed to better understand this phenotype.
ICAD: intracranial dissection
Arterial dissection involves the disruption of each layer of the arterial wall, which can lead to an intimal flap, intramural hematoma, pseudoaneurysm, and even artery rupture. Similar to intracranial atherosclerosis and MMD, intracranial dissection is reported more frequently in Asian patients [30]. Cervicocephalic arterial dissection is the leading cause of ischemic stroke in children and young adults. Most pathological studies on intracranial arterial dissection have revealed disruption of the internal elastic lamina and media, followed by subarachnoid hemorrhage. Hemorrhagic stroke can occur in up to 50%–60% of cases after intracranial dissection, but this could be an overestimate considering the selection bias in these studies, which were mainly conducted by neurosurgeons who are consulted only when there is evidence of subarachnoid hemorrhage [31,32]. Intracranial dissection without arterial rupture can cause ischemic stroke via several mechanisms. This includes luminal narrowing due to intramural hematoma, distal embolization of the thrombus formed within a false lumen, or occlusion of the perforating arteries by the dissected segment. A high clinical suspicion by a physician is warranted for a correct diagnosis. Due to the small caliber of the intracranial arteries, it is difficult to detect imaging clues for diagnosing arterial dissection using standard imaging tools [33]. Typically, several weeks after arterial dissection, the dissected segment is replaced by granulation tissue and subsequent neovascularization occurs in the thinned intima, potentially leading to a fusiform aneurysm or a healed vessel wall [32]. This necessitates caution when interpreting HR-VWI to evaluate ICAD, as discussed in the following section (Figure 2D).
ICAD: vasculitis
Inflammation of the cerebral arterial system is described as central nervous system (CNS) vasculitis [34]. A very large number of conditions are associated with CNS vasculitis, and the pathogenesis and mechanism of this disease entity is expansive. Although CNS vasculitis can manifest as large- and medium-vessel inflammation, which is easily detected on conventional lumen-based imaging techniques and HR-VWI, small arteries and leptomeningeal vessels can also be affected, which are beyond the imaging spatial resolution of conventional techniques. Thus, the negative predictive value of imaging for CNS vasculitis is low, and a negative imaging examination does not exclude the possibility of CNS vasculitis [35]. Several studies using serial imaging have reported that contrast enhancement of the vessels reflects vasculitic activity and is correlated with clinical outcomes in terms of recurrence and treatment effects (Figure 2E) [36-39].
On HR-VWI, CNS vasculitis may show periadventitial enhancement extending beyond the borders of the vessel wall itself in addition to concentric vessel wall thickening and enhancement [40]. This imaging feature may help distinguish between infectious CNS vasculitis and autoimmune/inflammatory CNS vasculitis and atherosclerosis. However, owing to the rarity of these infectious vasculitides, the discriminatory power of this imaging finding has not been well studied, and studies with larger sample populations are needed.
Go to :
Role of high-resolution vessel wall MR imaging in the evaluation of ICAD
As previously mentioned, the etiologic heterogeneity of ICAD is largely attributable to anatomical, histological, and pathologic differences between the intracranial and extracranial cerebral arteries. In particular, the intracranial location and small caliber of the intracranial arteries make it difficult to evaluate ICAD. To overcome these obstacles, strategies for visualizing the vascular walls of the intracranial arteries using HR-VWI have been developed. In this section, we discuss the factors that should be considered when interpreting HR-VWI findings by vascular neurologists caring for patients with ICAD.
Clinical considerations on the image acquisition of HR-VWI
Due to the high submillimeter spatial resolution achieved by current HR-VWI techniques, the detailed vessel wall anatomy and disease processes of the intracranial arteries can be resolved. The inner luminal diameter of middle cerebral artery (MCA) is typically 2 to 4 mm, and the wall thickness of a normal, healthy MCA ranges from 0.2 to 0.3 mm, which is smaller than the clinically achievable HR-VWI imaging voxel size (Figure 3) [41]. High magnetic field strength, preferably at 3 T, is recommended to achieve a high signal-to-noise ratio (SNR) with the recommended goal of 0.5-mm isotropic resolution [41]. To accurately track an intracranial artery, which is often tortuous, three-dimensional (3D) volumetric image acquisition is recommended to perform multiplanar reformats and visualize the artery in the orthogonal plane. However, it is worth noting that there are many different protocols and pulse sequence combinations using 3D versus 2D imaging for imaging the intracranial arteries. Suppression of blood and CSF signals is necessary and achievable on T1-weighted sequences. Multiple tissue weightings, with and without contrast agents, are recommended for characterizing intracranial vessel walls and their diseases. A proton density (PD) sequence is superior to a T1-weighted image as it achieves a better SNR to visualize the anatomy of the intracranial vessels; however, contrast enhancement may be less obvious, and signal intensities of the CSF and vessel wall are similar [41]. Postcontrast T1-weighted images can be used to characterize various wall pathologies by visualizing contrast enhancement, which may be interpreted as inflammation from atherosclerotic plaque, fibrosis from chronic pathology, or VV (Table 1).
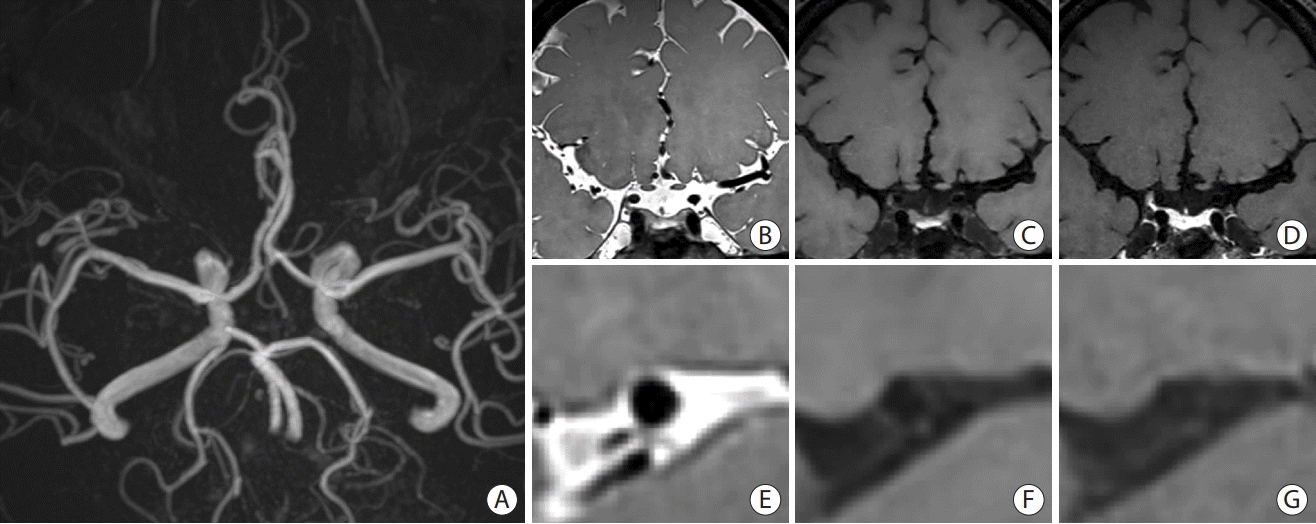 | Figure 3.A representative image of high-resolution vessel wall imaging of normal intracranial arteries. (A) Time-of-flight image. (B-D) Proton density (PD), T1-weighted, and T1-weighted with gadolinium (T1GD) coronal images showing the left middle cerebral artery (MCA). (E-G) PD, T1-weighted, and T1GD coronal image showing a cross-sectional view of the left MCA.
|
Table 1.
Imaging characteristics of vascular wall pathologies on different pulse sequences
![]()
The PD sequence has a high SNR and is used to measure the outer wall diameter [42,43]. Fibrotic tissue and lipid cores may appear as hyperintense signals on PD-weighted images. In T1-weighted images, the inner lumen and vessel wall are well differentiated; therefore, the luminal area can be identified [41,44]. T1-weighted imaging is also useful in identifying atherosclerosis because fibrotic tissue and the lipid core (without fat suppression) appear distinct with a hyperintense signal, and intraplaque hemorrhage appears hyperintense, especially in the subacute period [13,45]. Contrast-enhanced T1-weighted imaging also shows a well-demarcated inner lumen and vessel wall, with enhancement of the vessel wall due to inflammation, neovascularization, and VV. 41 T2-weighted imaging may reveal an intimal flap in dissection and perivascular edema in vasculitis with a hyperintense signal [41].
HR-VWI of intracranial arteries has several limitations. The first is the “slow-flow artifact,” which can cause vessel walls to appear falsely thick owing to inadequate blood signal suppression along the periphery due to slow laminar flow [46,47]. It is particularly ambiguous in certain vascular segments such as the Sylvian cistern near the MCA, dilated arteries, and petrous and cavernous ICA [46,47]. This artifact can be misinterpreted as arterial wall enhancement, and cross-checking with time-of-flight MR angiography (TOF MRA) and the pattern of the hyperintense signal is warranted [46,47]. Another interpretive pitfall is enhancement of the VV, which may mimic arterial wall thickening or vasculitis, especially in the presence of aging or atherosclerotic risk factors. Furthermore, the extracranial VV extends along the proximal intracranial segments of the ICA and VA immediately after dural penetration; this mild enhancement of the VV can be misinterpreted as vessel wall enhancement [41,46,47]. Finally, the venous plexus adjacent to the arteries may also be falsely interpreted as arterial wall enhancement [41]. For example, the petrous ICA is surrounded by the venous plexus of Rektorzik (e.g., the petrooccipital venous plexus) [48]. Ideally, these problems can be addressed by improving the spatial resolution and optimally suppressing blood and CSF flow, but often at the cost of increased acquisition time [46]. Efforts are underway to improve the resolution while reducing the acquisition time through the development of new sequences (e.g., motion-sensitized-driven equilibrium [MSDE] and delay alternating with nutation for tailored excitation [DANTE]), efficient imaging techniques (efficient k-space sampling trajectories), and hardware improvements [46,49-52].
Histological and pathological correlation of HR-VWI findings
Interpretation of HR-VWI findings should consider the clinical context of the patient’s laboratory and clinical examination findings. To date, there are few correlative radiology–pathology data, given the challenge of performing biopsies of diseased intracranial arteries. In lieu of pathology, serial HR-VWI can also add to our knowledge by revealing the presence of ICAD.
For example, a young ischemic stroke patient with a history of sudden severe headache may be clinically diagnosed with intracranial dissection. HR-VWI may reveal a hyperintense lesion on a precontrast T1-weighted image, which may be interpreted as an intramural hematoma. However, imaging findings are nonspecific, as vulnerable plaques with intraplaque hemorrhage or high lipid content may manifest similarly. Serial HR-VWI can help distinguish between intramural hematomas and atherosclerotic lipid components because subacute to chronic intramural hematomas show diminished T1 hyperintense signals as they become hypointense. In contrast, the high T1-weighted signal intensity of atherosclerotic plaques remain unchanged over time.
Postcontrast HR-VWI provides valuable diagnostic information. Strong vessel wall enhancement suggests vessel wall inflammation and may be a sign of an ongoing pathological process. However, because persistent enhancement can be caused by neovascularization in the fibrotic tissue, the enhancement itself lacks specificity, and the clinical history can add valuable context to these cases.
When interpreting HR-VWI results, it is necessary to check the intracranial arteries in the orthogonal plane to review the eccentricity of the lesion [41]. For example, atherosclerotic plaques are thought to predominantly show eccentric vessel wall thickening, whereas vasculitis tends to show concentric wall thickening [40]. Additionally, it is crucial to remember that normal or reactive findings can mimic pathological findings. For example, poor blood suppression due to slow intraluminal laminar flow can be misinterpreted as enhancement, particularly on postcontrast T1-weighted images because it appears slightly hyperintense on T1-weighted images. This pitfall can be especially problematic in branches distal to the M2 segment and arteries adjacent to the veins. The VV, which is sparse in the intracranial arteries, can develop with aging or disease processes and may appear to be slightly enhanced. This finding can mimic vasculitis or be mistaken for plaques, particularly in the proximal intracranial ICA or VA, where the VV is often present after dural penetration [53]. Finally, after thrombectomy during recanalization treatment using a stent retriever, the resulting endothelial injury may result in strong enhancement in the acute period [54].
Go to :
Utilization of high-resolution vessel wall MR imaging in clinical practice
The clinical context that necessitates additional HR-VWI after routine MR imaging and MRA may include (1) a high suspicion of non-atherosclerotic ICAD, (2) evaluation of ICAD activity, (3) diagnosis of branch occlusive disease without significant stenosis, (4) exclusion of symptomatic ICAD without detectable luminal narrowing on conventional imaging, and (5) follow-up of ICAD after treatment. Owing to the limited experience and lack of imaging-clinical-pathological correlations, the interpretation of HR-VWI findings requires careful inspection and clinical correlation. There is an overlap in HR-VWI characteristics across ICAD pathologies, as discussed in the following section. It is difficult to completely discern various intracranial pathologies on a single HR-VWI because there are many overlaps in their appearances. Thus, close communication between clinicians and radiologists is necessary for a reasonable evaluation.
Clinical interpretation of HR-VWI findings in ICAD
ICAD: atherosclerosis
When symptomatic ICAD is identified in a patient with atherosclerotic risk factors, such as advanced age, a long history of hypertension, diabetes, dyslipidemia, and smoking, vascular physicians typically first consider atherosclerosis as an etiology. It has been suggested that atherosclerotic ICAD does not share the same histopathological characteristics with extracranial arteries. Histological analysis has shown that intracranial atherosclerosis has more fibrous components and fewer necrotic lipid-rich cores than extracranial atherosclerosis [13,15]. Therefore, it is not easy to identify the typical imaging features of HR-VWI for atherosclerotic ICAD. Instead, strong enhancement on postcontrast T1-weighted images is a feature common to vasculitis and may be the sole feature visualized in ischemic stroke. In the correct clinical setting and history, HR-VWI can increase the diagnostic confidence of vascular physicians of atherosclerotic origin (Figure 4A). Intracranial atherosclerotic plaque characteristics can differ, leading to differences in the mechanisms of stroke. For example, intracranial atherosclerotic plaques can develop and progress by “creeping” along the superior/dorsal MCA vessel walls, leading to occlusion of the lenticulostriate arteries and ischemia. In contrast, intracranial atherosclerotic plaques may progress with vulnerable features, leading to plaque rupture, thrombus development, and embolism. Prior work has described this as branch-occlusive (B)-type versus coronary (C)-type atherosclerosis [55]. With HR-VWI, identifying B- versus C-type atherosclerosis can add precision to diagnosis, as these two different types of plaques may warrant different tailored therapies to reduce the risk of recurrent stroke.
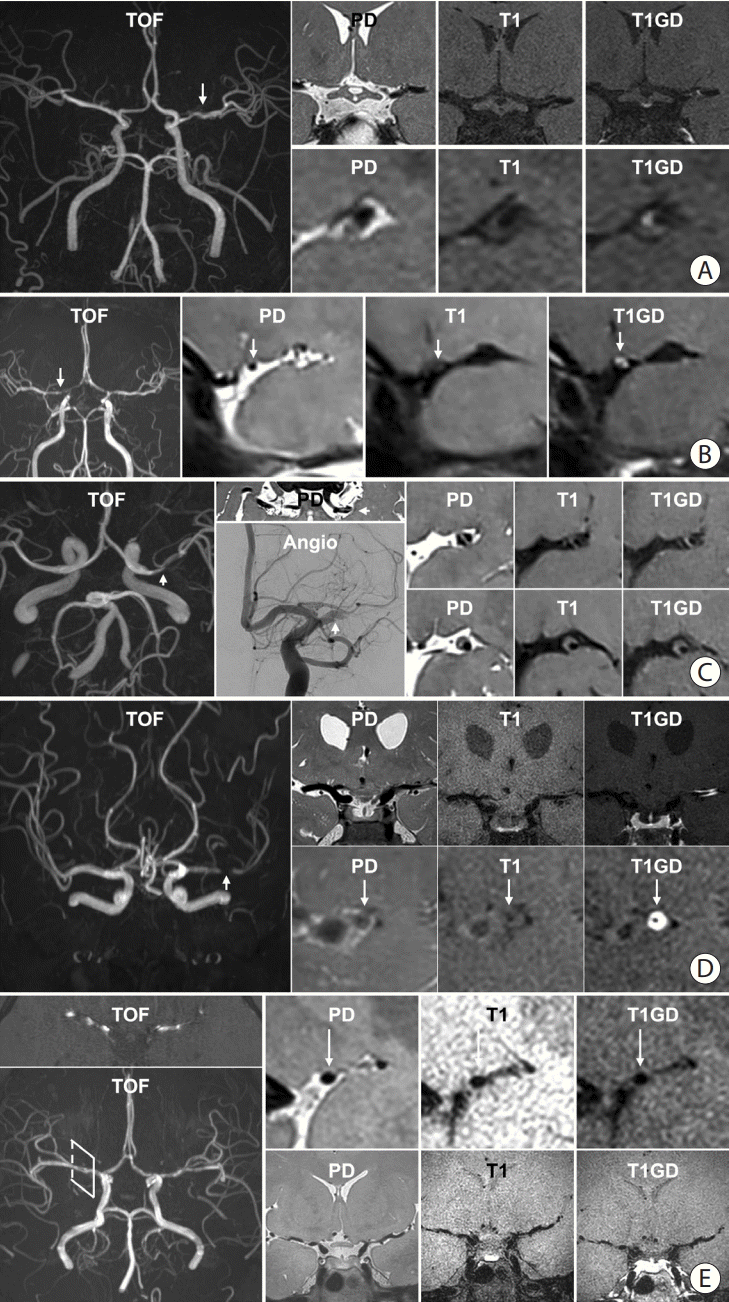 | Figure 4.Representative high-resolution vessel wall imaging (HR-VWI) images of various intracranial arterial diseases. (A) HR-VWI findings of atherosclerosis in a 45-year-old male with prehypertension and hypercholesterolemia. A stenotic portion (arrow) at the left middle cerebral artery (MCA) is identified on the time-of-flight (TOF) image. The proton density (PD) image shows no total vessel wall diameter stenosis. The T1 image shows eccentric vessel wall thickening with isointense signal suggesting a lipid core or fibrous cap. The area appears to be enhanced on the T1-weighted with gadolinium (T1GD) image. (B) HR-VWI in a 35-year-old female with moyamoya disease presenting with right MCA infarction. A heterozygous RNF213 c.14429G>A mutation was found. The TOF image shows stenosis at both distal internal carotid artery (ICA) to proximal A1 and M1. The arrow indicates the most stenotic part of the right MCA. Concentric wall thickening involving the right MCA with enhancement and negative remodeling was noted on PD, T1, and T1GD images. (C) HR-VWI of a left MCA dissection in a 48-year-old male who underwent endovascular treatment due to a left MCA infarction 4 months earlier. TOF-magnetic resonance angiography (MRA) shows a stenotic portion of the left MCA (arrow). The intimal flap is clearly visible on the PD axial image. Angiography at the time of endovascular treatment shows the intimal flap (arrow). PD, T1, and T1GD sagittal images show a double lumen and an intramural hematoma. A lesion with hyperintense PD and hyperintense T1 signal and no enhancement suggests a late subacute stage of the intramural hematoma. (D) HR-VWI in a 47-year-old female with vasculitis. TOF image shows a severely stenotic portion of the left MCA (arrow). PD, T1, and T1GD coronal images show wall thickening and prominent enhancement of the left M1. Sagittal images show concentric wall thickening with negative remodeling with strong enhancement. The stenosis later progressed despite steroid treatment. (E) HR-VWI in a 27-year-old female with reversible cerebral vasoconstriction syndrome presenting with severe headache. TOF-MRA axial image shows multifocal stenoses of both MCAs. Reconstructed TOF-MRA shows multifocal mild to moderate stenoses at the distal ICA, MCA, anterior cerebral artery, and basilar artery. Sagittal images of the right MCA show normal appearance of the vessel wall. Coronal images show stenotic segments without any abnormal vessel wall signal. Intracranial vessels were significantly improved on follow-up HR-VWI.
|
ICAD: MMD or RNF213 spectrum disease
Little is known about the HR-VWI findings in MMD or RNF213 spectrum diseases. Clinically, they can be suspected in the presence of ICAD in adolescent or early adult cases of East Asian origin who have few or no atherosclerotic risk factors. Distal ICA or MCA occlusion with abundant basal collateral channels may leave little doubt about the diagnosis of MMD; however, severe isolated MCA stenosis or occlusion without basal collateral channels in an asymptomatic patient presents a diagnostic challenge [22,25]. Depending on the geographic origin of the patient, identification of RNF213 variants, if available, would be helpful for diagnosis.
On HR-VWI, MMD is characterized by strong enhancement in the distal ICA and extension into the proximal M1 or A1 [56]. In general, it exhibits concentric enhancement. However, in patients with an RNF213 variant, eccentric enhancement may be observed when accompanied by intracranial atherosclerosis [25,57]. On PD-weighted imaging, the outer diameter of the artery tends to decrease gradually as negative remodeling progresses. Rarely observed hyperintense lesions on precontrast T1-weighted images suggestive of atherosclerosis or dissection make it difficult to differentiate patients with RNF213 spectrum disease from those with vasculitis invading the MCA using HR-VWI imaging alone (Figure 4B).
ICAD: intracranial dissection
The presence of an intimal flap, intramural hematoma, and double lumen on the image strongly suggests dissection [33]. Even without imaging evidence, intracranial dissection cannot be ruled out because of the limited spatial resolution of current HR-VWI acquisition parameters and the variable severity of the disease [33]. Clinically, patients with ICAD who are young adults, males, current smokers, and Asian who present with a sudden headache upon exertion or in an eccentric posture should be screened for arterial dissection. Intracranial dissection can show either a concentric or an eccentric location with either longitudinal or focal segment involvement. In cases of acute hematoma formation, precontrast T1-weighted images may display a characteristically strong hyperintense signal with limited perilesional enhancement due to a weak inflammatory process in the vascular wall or the absence of neovascularization during the early period. It is essential to remember that the dissection itself results in an active vessel wall remodeling process, and the HR-VWI appearance varies based on the duration of the injury. This variable appearance is one of the reasons why dissection is a challenging imaging-based diagnosis (Figure 4C).
ICAD: vasculitis
Vasculitis is a descriptive term for a heterogeneous disease that causes an inflammatory response in vessel walls. As a result, clinical presentation and HR-VWI findings vary depending on the cause. In general, vasculitis shows concentric and avid vessel wall enhancement during the active disease period [40]. This concentric enhancement in vasculitis is different from that in MMD in that it is not typically accompanied by negative remodeling [43]. Unless disease activity is modified, vasculitis typically progresses to the subacute phase. In the long term, normal or slight improvement is observed if inflammation is successfully controlled (Figure 4D) [37].
ICAD: reversible cerebral vasoconstriction syndrome
Reversible cerebral vasoconstriction syndrome (RCVS) can manifest as ICAD. Patients with RCVS typically present with a dramatic emotional upsurge and accompanying hyperventilation or headache, frequently in conjunction with the offending medication; therefore, a clinical assessment can strongly support this diagnosis [58]. In challenging cases, HR-VWI can be used to assess vessel wall enhancement of the narrowed arteries seen on MR, computed tomography angiography, or catheter cerebral angiography. The literature suggests that RCVS is an autonomic dysfunction with minimal to no vessel wall enhancement, which contrasts with inflammatory vasculitis (Figure 2F) [40]. However, a few cases of diffuse smooth vessel wall enhancement have also been seen in RCVS, raising the question of RCVS subtypes [59]. The hallmark of RCVS is the reversibility of vessel abnormalities with a relatively good prognosis for these patients (Figure 4E) [59].
Clinical significance of follow-up HR-VWI
Serial HR-VWIs in a patient with ICAD can provide valuable information (Table 2). An increase in plaque burden and enhancement ratio on follow-up HR-VWI is associated with an increased risk of recurrent stroke [60]. Culprit lesions tend to maintain their enhancement in stroke patients and serial HR-VWI can provide information on whether the lesion is likely to be the culprit [61]. Statin is known to decrease wall enhancement in intracranial atherosclerosis [62]. Antiplatelets and statins can reduce plaque burden in both non-culprit and culprit lesions [63]. Recently, a study showed a decrease in plaque enhancement after balloon angioplasty or drug-coated balloon angioplasty on follow-up HR-VWI, indicating plaque stabilization [64]. Another study showed that reduced enhancement on early follow-up HR-VWI after balloon angioplasty and stenting could predict fewer recurrent ischemic events [65].
Table 2.
Reports of follow-up HR-VWI for various ICADs
| Pathophysiology | Study | Patients | Number | Country | Follow-up | Temporal change in HR-VWI findings |
|---|---|---|---|---|---|---|
| Atherosclerosis | Chung et al. [62] | ≤7 d IS with atherosclerotic ICAD | 77 | Korea | 6 mo | After high dose statin: enhancement↓, wall area index↓, and stenosis degree↓ |
| Shi et al. [71] | <8 wk IS, stenosis >30% | 58 | China | 6.2 mo | Plaque burden progression in FU was associated with recurrent stroke | |
| Lee et al. [72] | Symptomatic atherosclerotic ICAD | 35 | Korea | 12–24 mo | Initial presence and enhancement of plaque was associated with stenosis aggravation | |
| Zhang et al. [73] | Symptomatic atherosclerotic ICAD+EVT | 45 | China | Pre- and post- procedure | Drug-coated balloon angioplasty reduced plaque enhancement (vs. bare balloon angioplasty) | |
| Kwee et al. [61] | Symptomatic atherosclerotic ICAD | 14 | USA | 3–6 mo (6) | Culprit lesion: enhancement not changed | |
| ≥6 mo (6) | Non-culprit lesion: enhancement↓ | |||||
| <3 mo (2) | ||||||
| Shen et al. [60] | Symptomatic atherosclerotic ICAD | 67 | China | 334 d | Recur (+) group: plaque features not changed | |
| Recur (-) group: stenosis ratio↓, plaque burden↓, enhancement ratio↓ | ||||||
| Meng et al. [64] | Symptomatic atherosclerotic ICAD+drug coated balloon angioplasty | 29 | China | <3 mo (4) | Balloon angioplasty: stenosis degree↓, plaque hyperintensity↓, wall enhancement↓ | |
| 3–6 mo (14) | ||||||
| >6 mo (11) | ||||||
| Lin et al. [63] | Symptomatic atherosclerotic ICAD | 87 | China | 8 mo | Culprit plaque: plaque length↓, maximum thickness↓, NWI↓, stenosis degree↓, plaque contrast ratio↓ | |
| Nonculprit plaque: NWI, stenosis degree↓ | ||||||
| Xiao et al. [74] | Symptomatic atherosclerotic ICAD | 29 | China, USA | 8 mo | Recurrent group: maximum wall thickness↑, plaque-wall contrast ratio↑, plaque enhancement ratio↑ | |
| Huang et al. [75] | Symptomatic atherosclerotic ICAD | 37 | China | 3, 6, 9, 12 mo | 12 mo of medical treatment: plaque burden↓, enhancement↓ | |
| 6 mo after balloon angioplasty: plaque burden↓ | ||||||
| Wu et al. [65] | Severe atherosclerotic ICAD+angioplasty and stenting | 24 | Taiwan | 24 h, 4.5 mo | HR-VWI ≤24 h can predict a 1-year outcome following intracranial stenting | |
| Dissection | Kwon et al. [76] | Dissection | 43 | Korea | 40 d | Extracranial dissection group: stenosis improved, residual wall enhancement↓, intramural hematoma↓ |
| Lee et al. [66] | <7 days IS | 17 | Korea | 3–12 mo | Hematoma disappears in 6 months. Most lesions healed in 12 months. | |
| MMD | Kim et al. [56] | Adult MMD | 66 | Korea | 30 mo | Negative remodeling occurred less in the cilostazol group (vs. other antiPLT vs. no antiPLT) |
| Lu et al. [67] | Adult MMD | 170 | China | 9.9 mo | Initial presence and progression of enhancement was associated with rapid stenosis progression. Vessel wall enhancement and rapid stenosis progression was associated with recurrent stroke. | |
| Vasculitis | Shimoyama et al. [38] | CNS vasculitis | 9 | Japan | 15.6 mo | Relapsed cases: enhancement↑ |
| Intensive immunosuppressive Tx: enhancement↓ | ||||||
| Cho et al. [36] | Varicella zoster vasculopathy | 1 | Korea | 2, 5 mo | After steroid and valacyclovir: enhancement↓ | |
| Song et al. [39] | Pediatric tuberculosis vasculitis | 1 | USA | 13, 21, 99, and 237 d | After anti-tuberculosis medication: leptomeningeal, wall, and parenchymal enhancement↓ | |
| Patzig et al. [37] | CNS vasculitis | 45 | Germany | 239.5 d | Enhancement (+): relapse↑ | |
| Kang et al. [77] | CNS vasculitis | 41 | China | 5.3 mo | Less enhancement: stenosis improved | |
| Yeo et al. [68] | Antiphospholipid syndrome | 8 | Korea | 110 d | Extremely high B2GPIs titer → diffuse narrowing↑ despite medical Tx | |
| RCVS | Choi et al. [69] | Atypical RCVS | 1 | Korea | 1 wk, 1 mo | After nimodipine: stenosis improved |
HR-VWI, high-resolution vessel wall imaging; ICAD, intracranial arterial disease; IS, ischemic stroke; FU, follow-up; EVT, endovascular treatment; NWI, normalized wall index; MMD, moyamoya disease; antiPLT, antiplatelet agent; CNS, central nervous system; RCVS, reversible cerebral vasoconstriction syndrome; B2GPIs, beta-2-glycoprotein I.
![]()
In a study investigating changes in intracranial dissection, intramural hematomas began to disappear within 6 months, and most damaged walls healed within 12 months [66]. This finding provides insights into deciding whether to discontinue antiplatelet therapy, especially in young patients, who are more susceptible to intracranial dissections. In MMD, a study showed that the progression of negative remodeling on follow-up HR-VWI was slower when cilostazol was administered than when other antiplatelet agents were used [56]. Another study showed that rapid progression of arterial stenosis increased the risk of recurrent stroke in patients with MMD [67]. In vasculitis, the clinical course and degree of enhancement are usually well correlated, and changes in follow-up HR-VWI can guide management to continue or taper steroids or other immunotherapies [36-38]. A recent case series revealed temporal changes in antiphospholipid syndrome-associated proliferative vasculopathy using serial HR-VWIs [68]. An atypical case of RCVS without typical thunderclap headache was also diagnosed using serial HR-VWI [69].
Determining the appropriate timing for follow-up HR-VWI is critical and should be etiology-specific. Our unpublished data (Kang DW, 2024) revealed that intracranial dissections exhibit notable changes in vessel wall morphology over a shorter time frame than atherosclerosis. We suggest a follow-up of approximately 3–6 months for dissection and 6–12 months for atherosclerosis. For RCVS, a 12-week follow-up would be reasonable in line with the diagnostic criteria [70]. MMD can be monitored for approximately 24 months to assess progression unless otherwise indicated [56]. For vasculitis, imaging should be performed when critical treatment decisions are pending and when a clinical–radiological correlation is needed. Therefore, follow-up imaging may be important for the diagnosis and treatment of ICAD, assessment of treatment response, and clinical decision-making.
Go to :
Conclusions
Intracranial arteries differ from extracranial cerebral arteries in several anatomical, embryological, and histological characteristics. In addition, ICAD has multiple etiologies, including atherosclerosis, dissection, moyamoya or RNF213 spectrum disease, and vasculitis. Advancements in HR-VWI techniques have enabled image acquisition at submillimeter spatial resolution, allowing for the visualization of the arterial wall and facilitating the diagnosis of vascular pathology with higher precision.
Currently, HR-VWI is predominantly used in clinical practice to differentiate between different disease types in ICAD. However, given the limited radiology-pathology data for ICAD, considering both the clinical context and timing of imaging from symptom onset is critical for HR-VWI interpretation. Different conditions within ICAD exhibit distinct temporal profiles, which impact imaging characteristics and may contribute to more accurate diagnoses over time. Nonetheless, there are limitations to qualitative interpretation, visualization of submillimeter structures, and spatial co-registration between image sequences in HR-VWI. Additional clinical studies with an adequate number of cases and extended follow-up durations are necessary to address these challenges. Advancements in image processing and deep learning techniques may offer solutions to overcome these limitations. Future research should focus on investigating the temporal profile of HR-VWI findings and developing quantitative interpretation approaches to enable precision in HR-VWI image-based decision-making.
Go to :
 XML Download
XML Download